
Single-cell RNA sequencing is giving engineers and scientists the ability to better understand and manipulate the smallest unit of a living organism — the single cell. This technique has already ushered in breakthroughs in biology and medicine. However, challenges and opportunities abound.
Single-cell RNA sequencing (scRNA-seq) is a technique used to simultaneously profile all expressed genes within a single cell (i.e., transcriptome-wide profiling). It involves various biological, chemical, and physical techniques to manipulate a minuscule amount of RNA from a single cell. The ability to obtain comprehensive gene-expression profiles at single-cell resolution has enabled researchers to rapidly identify and characterize novel and rare cell types, which in turn provides a foundation for the development of new therapeutic avenues — revolutionizing the pace of biological discovery and clinical innovation.
This article provides an overview of scRNA-seq technology and discusses various techniques for performing this type of sequencing. It also discusses emerging developments, opportunities, and challenges from an engineer’s perspective throughout.
Genomics at single-cell resolution
In 1959, when Richard Feynman first gave his famous lecture “Plenty of Room at the Bottom,” he conjectured that if we could understand and manipulate matter at the atomic scale, it would enable a myriad of previously unimaginable discoveries and innovations. Today, similarly profound advancements are happening in the life sciences field.
The single cell is the smallest functional unit of living beings, and bioengineers and biologists have long studied life at the single-cell level. But most approaches used in the past to characterize single cells — from physical observations of single cells to single-neuron electrophysiology measurements — have focused on just a few features, properties, or genes. Cellular RNA levels, which is a well-accepted proxy for cellular identity and behavior, could be measured for a panel of chosen genes by techniques such as micro-array analysis, but the ability to measure all gene expression in a cell and provide systems-level insights has, until recently, remained elusive.
The arrival of next-generation nucleic-acid-sequencing technologies drastically improved cellular readout (i.e., results) — from measuring selected gene targets to profiling all genes, or producing so-called whole transcriptomes (i.e., maps of all RNA transcripts for a given sample) (1). Initially, whole transcriptomes were generated from tissue samples, with all of the cells in that tissue blended together into a smoothie of sorts. But since each tissue in the body contains a multitude of cell types, each with diverse functions, a profile of a “tissue smoothie” is informative to a limited extent. The recent breakthroughs in transcriptome amplification, coupled with next-generation sequencing technology, culminated in the creation of highly sensitive whole-transcriptome analysis methods for single cells (2, 3).
In the first published scRNA-seq study on mammalian cells, Fuchou Tang and colleagues used scRNA-seq to investigate mouse cells during early embryonic development (4). The scientists used scRNA-seq to gain novel insights into the embryonic development process with unprecedented clarity, and, in the process, showed that scRNA-seq can overcome challenges related to analyzing small samples of biological material. Developmental biology, especially early-stage embryonic development, continues to be an area of research in which scRNA-seq is proving to be an indispensable tool for discovery.
Cancer treatment is another area of research that is benefiting greatly from scRNA-seq. Researchers have used scRNA-seq to unequivocally demonstrate and investigate the intratumoral heterogeneity in glioblastoma and colorectal cancer — shedding light on previously unknown mechanisms of cancer relapse and drug resistance, and exploring fundamental questions such as the validity of the cancer stem-cell model and the effects of immune interactions within tumor microenvironments (5–8).
A third important application of scRNA-seq is in the discovery and identification of new cell types, which has notably given rise to the Human Cell Atlas Project — a consortium established to characterize and catalog all distinct cell types in the human body in healthy and various disease states (9). The Human Cell Atlas Project, arguably the Human Genome Project of this era, is poised to have an equal or even greater impact on human health than its predecessor. The consortium has already identified novel cell types in the intestines, lungs, and many other tissue types, and has revised the taxonomy of dendritic cells in the immune system (10–13).
Many ways to skin a cat; many ways to sequence a cell
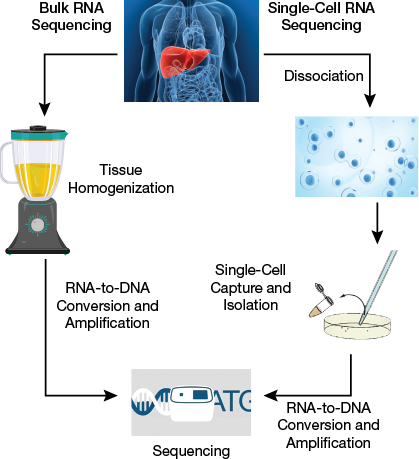
▲Figure 1. Bulk RNA sequencing maps the genome from tissue samples blended together into a bulk mixture. This method does not account for the differences between cells within a single tissue. Single-cell sequencing, on the other hand, sequences genetic material from one cell at a time, which provides more information about the cells within tissue samples.
RNA sequencing, whether for millions of cells or a single cell, follows a similar general procedure (Figure 1).
The main differences between the many-cells and single-cell methods are in the sample preparation and the RNA-to-DNA conversion chemistry. One of the challenges in sequencing single cells is the limited quantity of available RNA, which makes loss or damage of RNA during isolation and extraction extremely detrimental to the results. Cell viability is critical to the integrity of RNA-seq results, because cells respond to stress with changes in gene expression, or worse, RNA degrades if the cell begins to die. Therefore, dissociation of tissue samples into single cells and isolation of these single cells, without causing them too much trauma, are critical steps to preserving RNA integrity in the scRNA-seq workflow.
Dissociation. A combination of mechanical agitation and enzymatic digestion is typically used to release individual cells from a tissue matrix. The unique composition and structure of different tissue...
Would you like to access the complete CEP Article?
No problem. You just have to complete the following steps.
You have completed 0 of 2 steps.
-
Log in
You must be logged in to view this content. Log in now.
-
AIChE Membership
You must be an AIChE member to view this article. Join now.
Copyright Permissions
Would you like to reuse content from CEP Magazine? It’s easy to request permission to reuse content. Simply click here to connect instantly to licensing services, where you can choose from a list of options regarding how you would like to reuse the desired content and complete the transaction.