
Proteins could transform the chemical process industries if they could be engineered to facilitate the production of biofuels, molecular medicines, and other specialty chemicals. This reality is nearing as expanded knowledge of proteins and improvements in computational tools are making it possible to design specific protein structures and functions.
Proteins are vital to life on Earth: They replicate and repair DNA, ensuring the continuation of genetic material. They form internal support structures for cells and routes for intracellular transport of materials. And, perhaps most importantly, they catalyze a vast array of chemical reactions, speeding creation, organization, and consumption of important molecules.
Proteins are the most common catalysts nature uses to convert molecules from reactants to products. These protein catalysts function at room temperature and pressure and are often highly selective. Chemical engineers are resourceful and creative and have devised numerous methods of producing chemicals from feedstocks, yet many processes using traditional catalysts require extreme conditions that increase cost and reduce safety.
For many production processes, protein solutions have the potential to be less expensive, safer, and more effective than traditional catalysts. Although the utility of proteins has been recognized for a long time, they are still far from as prevalent as their potential suggests they could be. The reason: It is often too difficult and expensive to engineer a protein for a particular purpose.
Nearly all commercially meaningful efforts to engineer proteins have relied on experimental methods, which are limited to changing an existing protein. A sequence of 100 amino acids, for example, can be arranged into 20100 (~1.3×10130) proteins — significantly more variants than the number of electrons in the universe (1). There are many known proteins with more than 500 amino acids, making the number of possible proteins exceedingly vast.
Nature has provided only a minuscule fraction of possible protein structures, and it is effectively impossible to assemble a random protein with any function, let alone a specific function. Because the requirements of industrial applications are often far removed from natural conditions, it may be impossible to identify a protein candidate that can feasibly be modified.
Computational protein engineering overcomes some of the limitations of experimental methods by exploring protein folds not observed in nature and designing novel proteins from scratch (i.e., de novo design). Designing proteins at-will could be a powerful way to improve many chemical engineering processes. Although computational methods have had limited success, improved methods and expanded computational resources are rapidly increasing the areas in which these methods can be reliably applied.
This article discusses protein structures and computational engineering methods, provides examples to demonstrate state-of-the-art technologies, and examines the future of computational protein engineering.
Protein structures
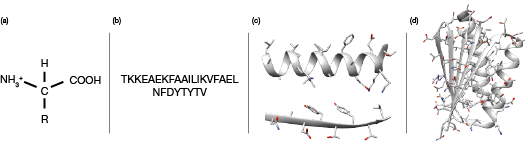
▲Figure 1. All proteins are composed of a linear chain of amino acids. (a) All amino acids have the same backbone structure, but they have different side chains bound to the central carbon. (b) A protein’s primary structure is the linear order of its amino acids. (c) Amino acids near one another in the primary structure assemble into secondary structure elements, such as α-helixes and β-strands. (d) The secondary structure elements fold into the overall complex protein fold, or tertiary structure, which brings into close proximity amino acids that may be far from one another in the primary structure.
Proteins are composed of 20 standard amino acids. The amino acids all share a similar structure (Figure 1a) — an amine group bound to a central carbon, to which a sidechain and a carboxyl group are also attached. The sidechains differentiate the amino acids.
A protein’s primary structure (Figure 1b) is the linear order of its amino acids, which are connected to each other by a peptide bond between the carboxyl group and the amine group. The primary structure is considered to have a direction, which is defined as starting with the amino acid that has an unbound amine group and ending with the amino acid that has an unbound carboxylic acid group.
Proteins do not exist as extended, linear conformations, but fold into complex, thermodynamically favorable geometries. Secondary structure elements (Figure 1c), such as α-helixes and β-strands, are arrangements of amino acids near one another in the primary structure.
The tertiary structure (Figure 1d) is the arrangement of the secondary structure elements into the overall complex fold. This involves one or more domains, which are stable sets of secondary structure elements.
Exceptions to these general protein structure guidelines exist. Some proteins incorporate rare amino acids, such as selenocysteine; some are modified by the addition of carbohydrate, phosphate, or other groups that affect their function or stability; and still others are intrinsically disordered and have dynamic tertiary and secondary structures. In general, however, protein structures and functions are determined by their primary, secondary, and tertiary structures. All experimentally determined protein structures are deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB), which currently contains more than 130,000 structures (2).
The complex structures of proteins are the result of finely balanced interplays between enthalpic and entropic forces. Computational methods use force fields to design and predict protein structures. Force fields, such as Amber (3), CHARMM (4), GROMACS (5) and Rosetta (6), are mathematical functions that calculate forces and energies between atoms, amino acids, and proteins. These calculations treat atoms as spheres of constant size and charge with fixed parameters for energy functions. Typical terms in the calculations include van der Waals forces, electrostatic forces, solvation effects, bond lengths, bond angles, and dihedral bond angles. Statistical parameters for the presence of naturally occurring features, such as length and angles of hydrogen bonds and the frequency of certain amino acids, are also used.
The most common types of calculations are molecular mechanics (MM), which predict the energy minimum of a protein structure, and molecular dynamics (MD), which calculate the behavior of the protein over time. The fixed-parameter approximations of MM and MD can cause inaccurate predictions. Density functional theory and quantum mechanics calculations do not involve these approximations and are sometimes used to improve accuracy, but they require...
Would you like to access the complete CEP Article?
No problem. You just have to complete the following steps.
You have completed 0 of 2 steps.
-
Log in
You must be logged in to view this content. Log in now.
-
AIChE Membership
You must be an AIChE member to view this article. Join now.
Copyright Permissions
Would you like to reuse content from CEP Magazine? It’s easy to request permission to reuse content. Simply click here to connect instantly to licensing services, where you can choose from a list of options regarding how you would like to reuse the desired content and complete the transaction.