
Scientists must learn how to open and manipulate chromatin to gain complete access to eukaryotic DNA for genome engineering.
For decades, scientists have used DNA-binding enzymes derived from bacteria to artificially edit DNA sequences in eukaryotic hosts, including human cells. Modern technologies have enabled scientists to better understand and manipulate the chromosomal structures that impede or enhance access to the underlying DNA.
The genomic DNA of eukaryotic cells is packaged within the nucleus as a protein-rich complex known as chromatin. Chromatin dynamically regulates gene expression by locally altering the density of DNA compaction and modifying associations with chromatin remodeling factors. Tightly compacted areas of chromatin are known as heterochromatin. The DNA within heterochromatin is inaccessible to endogenous transcription factors, as well as small-RNA-guided nucleases (e.g., CRISPR/Cas9) and protein-DNA binding nucleases (e.g., transcription activator-like effector nucleases [TALENs] and zinc finger nucleases [ZFNs]). Gaining complete access to the entire genome is an important step toward realizing the full potential of epigenomic and genomic engineering.
To this end, recent studies have investigated several key questions: What specific features of chromatin impact DNA accessibility to endogenous and exogenous effectors? How do these features alter stable engagement of gene-editing enzymes with their targets? And, how can we manipulate chromatin to overcome inhibition of gene-editing activity?
Transcription can be initiated within condensed regions of the genome, as evidenced by the chromatin reorganization that accompanies gene expression reprogramming in stem cells. This phenomenon suggests that scientists can harness endogenous machinery to gain access to DNA regions of interest. Scientists have recently begun to develop site-specific approaches to precisely manipulate local chromatin states at specific genes using this machinery.
This article reviews the structural elements of chromatin and how they affect gene-editing efficiency, as well as the few studies that have attempted to improve DNA editing in heterochromatin. It then discusses the methods employed by cells to open chromatin and reviews the mechanism behind each method, its potential to be engineered, and previous attempts to do so. Finally, the article examines the remaining challenges to opening chromatin for genomic engineering, as well as important design parameters that warrant further investigation.
Closed chromatin is a barrier to genome editing
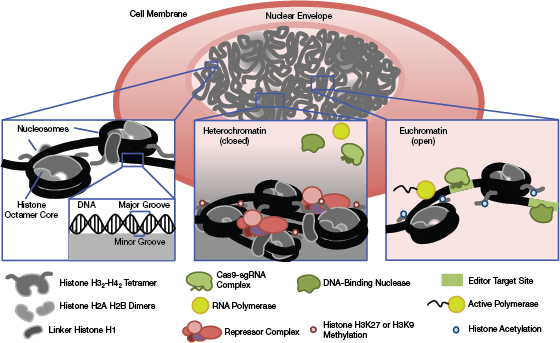
▲Figure 1. The core structure of chromatin is the nucleosome, an octamer of distinct histone proteins, around which DNA is wound. Highly compacted areas of chromatin are characterized by the localization of repressor complexes, repressive histone tail modifications, and linker histones that bring adjacent nucleosomes into close contact. This dense structure creates a liquid-like compartment through phase separation that precludes the interaction of DNA binding proteins (such as editing endonucleases) with DNA. Loosely packaged chromatin or euchromatin allows for active transcription of DNA and interaction with binding factors. Histone tail acetylation in euchromatin recruits these associated factors.
The primary structural unit of chromatin is the nucleosome. Nucleosomes are composed of DNA surrounding an octamer of histone proteins (Figure 1). Arrays of nucleosomes can assemble into higher-order structures through local interactions, such as binding of linker histones near the nucleosome dyad axis or co-localization of repressor complexes (1). These structures may be further organized and compacted through interactions with distant genomic domains through liquid-phase separation and bundling of chromosomal structures via interactions with the nuclear envelope (1, 2).
DNA at the histone-octamer interface is shielded from the nuclear environment. Furthermore, bending of the DNA around the histone complex distorts the width of the major groove in the double helix, resulting in a suboptimal fit for ZF proteins that interact with B-form DNA (3). Nucleosome positioning dynamically regulates the genome. Dense local nucleosome occupancy on the chromatin fiber prevents transcription factors (TFs) and other DNA binding proteins involved in transcription, replication, and repair from accessing DNA.
In assays where nucleases are used to cut DNA within whole nucleosomes, high levels of cleavage reproducibly distinguish open chromatin regions, whereas closed chromatin regions show low levels of cleavage (4). Physical interactions between endogenous nuclear proteins and DNA block the nucleases from accessing the DNA, which further reinforces the concept that access to DNA is influenced by chromatin structure.
Chromatin prevents gene-editing enzymes from stably engaging with their targets
Since their discovery in the 1970s, prokaryotic proteins that bind to specific DNA sequences and carry out site-specific DNA cleavage — e.g., Type II restriction endonucleases (5) — have inspired the development of customizable nuclease-based systems that cut and alter DNA sequences in living eukaryotic cells. Among these is the newcomer Cas9. This genetic editing tool is particularly attractive because only a short (~20 base pair [bp]) single-stranded guide RNA (sgRNA) is required to direct the Cas9 endonuclease to a specific location in the genome. Synthetic biologists and cell engineers have designed many alternate CRISPR-Cas9 systems that harness the highly specific and customizable binding properties of Cas9-sgRNA complexes.
Cas9 binding and endonuclease activity can be disrupted by dense nucleosome occupancy in vitro and in vivo (6–8). The link between chromatin inaccessibility and Cas9 inhibition has been demonstrated in mice, humans, and zebrafish (9). While other nucleases such as TALEs may also be perturbed by heterochromatin (10), Cas9-based systems show a pronounced variability in robustness across genomic loci in different eukaryotic models. Variability in Cas9 editing can be linked to variance in chromatin architecture across the genome (11). The context dependence of Cas9 editing must be overcome for Cas9 to be the broadly effective, flexible tool genomic engineers require.
To this end, scientists have investigated CRISPR-Cas9 activity within chromatin to identify critical design parameters for editing efficiency. The primary design element of any CRISPR-Cas9 system is the sgRNA. In vivo studies have shown that sgRNA selection impacts editing efficiency in heterochromatin (9, 12) and that sites with increased editing efficiency are often positively associated with areas of active transcription or open chromatin (12). The distance between nucleosomes and the sgRNA recognition site has been shown to impact editing (7) — areas with dense nucleosome occupancy are more likely to exhibit variability in editing across nearby target sites.
Cas9 protein concentration may also be an important parameter for improving editing in heterochromatin. Sampling of DNA binding sites by the Cas9-sgRNA complex is slowed in heterochromatin, suggesting that increasing protein concentration may increase sampling and successful binding events (13). Likewise, editing is also delayed in heterochromatin in a concentration-dependent manner (14).
A few attempts have been made to engineer the limited intrinsic chromatin-opening ability of Cas9 proteins. Barkal et al. report that a catalytically dead Cas9 (dCas9) can induce chromatin accessibility as measured by DNAse (i.e., a nuclease that degrades DNA) hypersensitivity and gene activation at 16 previously inaccessible genomic loci in mouse embryonic stem cells (mESCs) (15). However, previous work shows that Cas9’s ability to open chromatin is highly dependent on local chromatin structure, which was not investigated through either the interrogation of histone translational modifications (PTMs) or nucleosome positioning in Barkal et al. Their dCas9 system was also genomically integrated, possibly yielding higher dCas9 expression levels than by transient transfection.
Another group, Chen et al., used SpCas9 derived from the bacteria Streptococcus pyogenes to assist in opening chromatin for FnCas9, a novel RNA-guided nuclease (16). Using this approach, multiple SpCas9s were targeted to sequences 7 bp and 50 bp away from the FnCas9 target, presumably forcing open local chromatin, although alterations to chromatin structure were not determined in their study. This approach does improve FnCas9 function, but the mechanism and efficacy across cell types and loci remains unclear.
Although Cas9 design parameters can be manipulated in some contexts, inhibitory nucleosome occupancy remains an obstacle in many cases. Guide RNA target sites may be limited for certain applications and, although high concentrations of Cas9 may be able to surmount chromatin inhibition (14), this may be both difficult to achieve and undesirable in vivo, where the host immune response to Cas9 must be taken into account (17). Ultimately, we need to remodel local chromatin structure to reduce variability and increase efficiency of Cas9-mediated editing throughout the genome.
Manipulating chromatin to overcome inhibition of gene-editing activity
Within the dense throng of chromatin, how do TFs and other DNA-binding proteins ultimately find their binding domains? The compacted nature of the eukaryotic genome is a dynamic physical barrier to any process that requires access to DNA.
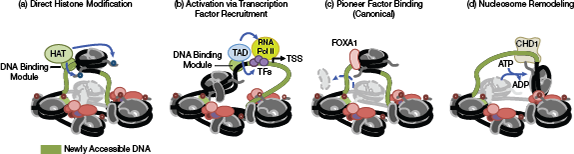
▲Figure 2. (a) Targeting of direct histone modifiers, such as histone acetyltransferase (HAT), to specific sites allows activation-associated histone tail modifications to be locally deposited, inducing the opening of chromatin. (b) Site-directed activators induce chromatin opening and the activation of transcription by recruiting additional transcription factors and polymerases. TAD = transcriptional activation domain; TSS = transcription start site. (c) FOXA1 is shown as an example of a pioneer factor with structural homology to linker histones; this homology enables disruption of condensed nucleosome organization and increased DNA accessibility. (d) Chromatin remodelers, such as CHD1, directionally shift, slide, or replace nucleosomes to modulate local chromatin accessibility. The DNA binding module represents any peptide (e.g., zinc finger or TALE) or complex (e.g., deactivated Cas9-gRNA) that can be fused to an effector module (e.g., HAT or TAD) to target it to a specific location in the genome.
Nucleosome positioning is regulated by several factors that determine local accessibility. Nucleosomes bound to DNA can be displaced by ATP-dependent chromatin remodelers (CRs) or certain classes of pioneer transcription factors (PFs) that interact with both other TFs and local histone modifications (18). Nucleosome displacement plays a role in promoter activity and gene expression by exposing DNA to regulatory factors. Thus, site-specific nucleosome depletion is an attractive option for opening chromatin to enhance artificial, nuclease-mediated editing. The next sections describe the native mechanisms of chromatin opening (Figure 2), previous attempts at harnessing the cellular mechanisms that may assist in engineering chromatin (Table 1), and their potential to generate Cas9-accessible chromatin states.
| Table 1. Many research groups have studied chromatin-opening mechanisms. | |||||
| Domain | General Function* | Effect(s) on Chromatin | DNA Binding Domain(s) | Model... | |
Would you like to access the complete CEP Article?
No problem. You just have to complete the following steps.
You have completed 0 of 2 steps.
-
Log in
You must be logged in to view this content. Log in now.
-
AIChE Membership
You must be an AIChE member to view this article. Join now.
Copyright Permissions
Would you like to reuse content from CEP Magazine? It’s easy to request permission to reuse content. Simply click here to connect instantly to licensing services, where you can choose from a list of options regarding how you would like to reuse the desired content and complete the transaction.