
This article highlights developments in single-cell epigenomic monitoring and modulating tools, as well as advances in synthetic epigenetic circuits. These tools are used in important biomedical and industrial applications.
Heritable and reversible chemical modifications of chromatin, i.e., histone post-translational modifications and DNA methylation, can regulate gene expression and alter cell phenotype without changing the underlying DNA sequence. Histone and DNA modifications include methylation, acetylation, phosphorylation, and ubiquitylation, among others. Epigenetic writers and erasers selectively introduce and remove epigenetic modifications. For example, the histone methyltransferase (writer) can transfer a methyl group from S-adenosyl methionine (SAM) onto specific lysine residues on histones, while the Jumonji domain-containing histone demethylase (eraser) can remove histone methylation via oxidation reactions.
Unlike genetic mutations, which are nearly permanently embedded in the genome and, thus, fairly resistant to external stimuli, epigenetic modifications are dynamic and can be modulated by a variety of factors. Exposure to external stimuli, such as chemicals in the environment (e.g., heavy metals and herbicides), oxidative stress (e.g., reactive oxygen), metabolites (e.g., 2-hydroxyglutarate and acetyl-CoA), and drug treatments, as well as alterations to the microenvironment (e.g., extracellular matrix [ECM]), can lead to changes in epigenetic modifications that can transiently or permanently alter the epigenome, causing different phenotypic behavior.
The dynamic nature of the epigenome provides both opportunities and challenges. Engineered small-molecule drugs and protein toolboxes are used to modulate and reprogram the epigenome without altering underlying genomic codes. These epigenetic engineering tools provide unprecedented opportunities to reprogram the transcriptional state of cells with high precision and resolution. The dynamic nature of epigenetic features, however, makes them difficult to track due to their intrinsic heterogeneity. Numerous factors, such as cell cycle phases, differentiation stages, cell ages, and varying locations within the ECM, can give rise to variations in epigenetic contents, making tracking single-cell epigenomics a difficult task.
This article covers recent developments in single-cell epigenomic monitoring and modulating tools and touches on some recent developments in synthetic epigenetic circuits. It also presents applications of these tools in important biomedical and industrial applications.
Monitoring epigenetic modifications
Epigenetic modifications are conventionally quantified via antibody-based assay via enzyme-linked immunosorbent assay (ELISA). ELISA uses antibodies that have a high binding affinity to the modification of interest to quantify the modification levels via colorimetric or fluorescent assays. Kits to quantify global epigenetic modifications are commercially available and can provide a relatively accurate picture of the changes in global epigenetic modification levels.
Gene-specific epigenetic modifications are typically characterized through bisulfite (BS) and chromatin immunoprecipitation (ChIP) approaches. Gene-specific DNA methylation is routinely quantified via BS-based approaches, which take advantage of a bisulfite-mediated deamination reaction that converts unmethylated cytosine to uracil while leaving methylated cytosine intact. The groups treated and untreated with bisulfite can then be analyzed via quantitative polymerase chain reaction (qPCR) (i.e., methylation-specific PCR [MSP]) or next-generation sequencing (NGS) to reveal the methylation level of cytosine at specific locations.
Gene-specific histone modifications are characterized by first enriching sequences containing the modification of interest through antibody-based pulldown assay of fragmented chromatins. This process, also known as ChIP, is typically followed by qPCR or NGS to quantify the modification level at selected loci or genome-wide, respectively.
The BS and ChIP approaches provide a robust assessment of the epigenome and are used in the Encyclopedia of DNA Elements (ENCODE) project, but they have several major drawbacks. The assays require a large number of cells: about 105 cells are needed to perform MSP, 3×103 for BS-seq, 2×105 for ChIP-qPCR, and 106 for ChIP-seq (1–3). ChIP-based analyses require a significantly larger number of cells because the precipitation step has a low recovery efficiency. In addition to requiring large numbers of cells, these assays are performed using cell extracts, so no information can be obtained regarding the distribution of epigenetic changes in the cell nucleus. The assays are also not compatible with live cells, so they cannot be used to monitor epigenetic changes in situ. However, significant progress has been made in recent years to address these concerns.
Single-cell BS-seq and ChIP-seq approaches have also been developed. Single-cell BS-seq uses drop-based technology to isolate single cells and characterize DNA methylation (4). The bisulfite reaction is carried out prior to installing sequencing adaptors to prevent loss of DNA information during bisulfite treatments. This process is followed by amplification reactions using random priming and then ligation to sequencing adaptors.
Single-cell ChIP-seq uses a similar technology to isolate individual cells (5). A microfluidic device enriches genomic DNA containing the histone modification of interest and performs sequencing reactions. Although in theory these techniques could be adopted for analyzing individual cells, a pool of about 100 cells is still required to generate NGS data with adequate coverage (5). Despite their low sequence coverage, single-cell BS-seq data accurately quantify DNA methylation in up to 48.4% of CpG (a cytosine nucleotide followed by a guanine) islands, and single-cell ChIP-seq can capture histone modifications in thousands of promoters (4, 5).
Single-molecule epigenetic analysis has also been demonstrated (6). High-throughput single-molecule imaging has been used in conjunction with single-molecule sequencing to reveal the different combinations of modifications of individual nucleosomes. This provides unprecedented resolution, revealing multiple epigenetic changes at a single location.
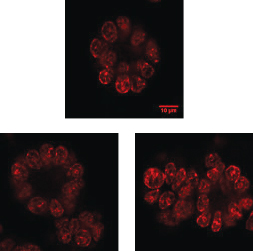
▲Figure 1. These typical immuno-stained images of breast cancer cells that are grown on Matrigel were collected using a confocal microscope with 60× magnification.
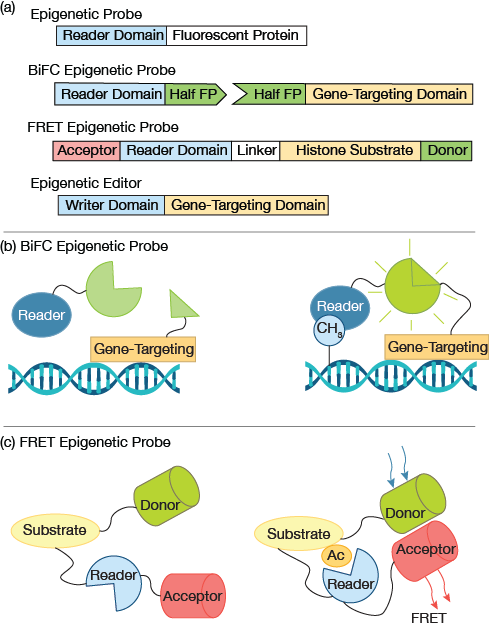
▲Figure 2. (a) A simple fluorescent epigenetic probe uses an epigenetic reader domain fused to a fluorescent protein tag. Bimolecular fluorescence complementation (BiFC) epigenetic probes employ split fluorophores, in which each half of the fluorophore is fused to an epigenetic reader domain and a gene targeting domain. A Förster resonance energy transfer (FRET) epigenetic reporter probe joins the reader domain and histone substrate with a flexible linker. A pair of donor and acceptor fluorophores are fused to the substrate and reader domain, respectively. A typical epigenetic editor contains an epigenetic writer domain fused with a gene-targeting domain, e.g., dCas9. (b) In a BIFC probe, when the gene-targeting domain and epigenetic reader domain bind in close proximately, e.g., a gene rich in the modification of interest, the halves of the fluorescent proteins complement each other and generate a fluorescence signal. (c) In a FRET probe, when the histone substrate contains the modification of interest, the fused reader domain recognizes the modification and triggers a conformational change, resulting in high FRET. No FRET signal is expected in the absence of the modification of interest.
Quantitative histology studies reveal cellular distribution of epigenetic patterns. Specifically, cells that are immuno-stained can be observed using high-resolution imaging (Figure 1). Collectively, the images provide snapshots of cellular epigenetic patterns in cells with no dynamic details. In addition, live-cell-compatible epigenetic sensors have been developed based on engineered epigenetic reader domains (Figure 2a) (7, 8). The engineered reader domains can be fused to fluorescent proteins or protein tags. Fluorescent images of sensor-transfected cells show the abundance of specific epigenetic modifications within individual cells.
Recent research takes this work further by combining epigenetic sensors with state-of-the-art gene-targeting tools, i.e., clustered regularly interspaced short palindromic repeats (CRISPR) and transcription activator-like effectors (TALEs) (9). The epigenetic reader and gene-targeting domain each contains half of a fluorescent protein (Figure 2a) that complement each other when in close proximity (Figure 2b). Fluorescent signals that arise from bimolecular fluorescence complementation (BiFC) can then be used to quantify gene-specific epigenetic changes within cells.
Epigenetic sensors that are based on reader domains are live-cell-compatible and, thus, suitable for cell tracking. Fab-fragment probes and Förster resonance energy transfer (FRET) probes have also been developed to query epigenetic changes in live cells. Antigen-binding fragments (Fabs that are about 50 kDa) are much smaller than antibodies and can be fluorescently tagged. They can be expressed in situ or readily diffused into cell nuclei with minimal perturbations to cell physiology. Multiple sensors for histone modifications, i.e., H3K9me2, H3K27me3, and H3K27ac, have been developed (10).
FRET reporters for epigenetic changes have been made for quantifying various histone modifications, including H4 acetylation and H3K9/K27 methylation (11, 12). FRET reporters have a modular design and are relatively easier to produce than Fab-fragments. A typical epigenetic FRET reporter consists of a pair of donor and acceptor fluorophores (e.g., fluorescent proteins), a modification-recognition domain (e.g., epigenetic readers), a flexible linker, and a substrate domain (e.g., core...
Would you like to access the complete CEP Article?
No problem. You just have to complete the following steps.
You have completed 0 of 2 steps.
-
Log in
You must be logged in to view this content. Log in now.
-
AIChE Membership
You must be an AIChE member to view this article. Join now.
Copyright Permissions
Would you like to reuse content from CEP Magazine? It’s easy to request permission to reuse content. Simply click here to connect instantly to licensing services, where you can choose from a list of options regarding how you would like to reuse the desired content and complete the transaction.