
(86d) Accelerated Saddle Point Refinement with Sella
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Computational Molecular Science and Engineering Forum
Recent Advances in Multiscale Methodologies
Monday, November 16, 2020 - 8:45am to 9:00am
In a recent publication[1], we describe a FOSP refinement procedure that accelerates convergence to a saddle point using iterative Hessian diagonalization. The Hessian-vector products that have been evaluated during diagonalization are used to construct a highly accurate approximate Hessian. In the full-diagonalization limit, the approximate Hessian constructed in this way becomes exact. The total number of diagonalization steps performed is controlled by a single convergence parameter, γ. Tighter convergence yields a more accurate Hessian – thereby reducing the number of refinement steps needed to locate the FOSP – at the cost of additional diagonalization iterations. The cost of diagonalization iterations can be balanced against the cost of additional geometry refinement steps by carefully tuning the value of γ.
Our method has been implemented in an open source software package, Sella[2]. Sella is written in Python and is compatible with all major operating systems and computer architectures. Sella uses the ASE[3] library to interface with a wide range of software packages for PES evaluations, including electronic structure theory packages and classical force-field packages. In particular, we have used the ASE interface to LAMMPS[4] for several test systems.
We measure the performance of our approach as implemented in Sella on two FOSP refinement benchmarks from optbench.org[5]. On one benchmark, we see an average reduction of over 50% in the number of gradient evaluations (consisting of diagonalization iterations plus geometry refinement steps) required to converge to a FOSP. On a second benchmark, we see an average reduction in the number of gradient evaluations of over 25%. Additionally, our implementation of iterative diagonalization requires an average of 30% fewer iterations to converge to the leftmost eigenvector of the Hessian compared to the best performing codes listed on optbench.org. When an approximate Hessian is available, our Jacobi-Davidson-based diagonalization routine requires on average 40% fewer iterations to converge than non-preconditioned Lanczos. These performance improvements are a direct result of the increased accuracy of the approximate Hessian matrix that our method enables.
[1] Hermes, E.D., Sargsyan, K., Najm, H.N., Zádor, J. J. Chem. Theory Comput. 15, 6536-6549 (2019).
[2] Hermes, E.D. Sella, 2019, DOI: 10.5281/zenodo.3379094.
[3] Larsen, A.H., Mortensen, J.J., Blomqvist, J., Castelli, I.E., Christensen, R., Dułak, M., Friis, J., Groves, M.N., Hammer, B., Hargus, C., Hermes, E.D., Jennings, P.C., Jensen, P.B., Kermode, J.B., Kitchin, J.R., Kolsbjerg, E.L., Kubal, J., Kaasbjerg, K., Lysgaard, S., Maronsson, J.B., Maxson, T., Olsen, T., Pastewka, L., Peterson, Al., Rostgaard, C., Schiøtz, J., Schütt, O., Strange, M., Thygesen, K.S., Vegge, T., Vilhelmsen, L., Walter, M., Zeng, Z., Jacobsen, K.W. J. Phys.: Condens. Matter 29, 273002 (2017).
[4] Plimpton, S. J. Comput. Phys. 117, 1 (1995).
[5] Chill, S.T., Stevenson, J., Ruehle, V., Shang, C., Xiao, P., Farrell, J.D. and Wales, D.J. J. Chem. Theory Comput. 10, 5476 (2014).
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, as part of the Computational Chemistry Sciences Program (Award Number: 0000232253).
Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. The views expressed in this article do not necessarily represent the views of the U.S. Department of Energy or the United States Government.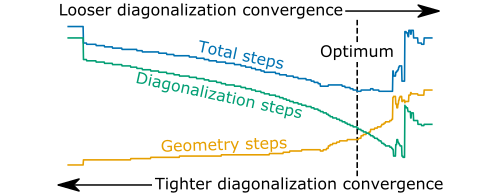
Checkout
This paper has an Extended Abstract file available; you must purchase the conference proceedings to access it.
Do you already own this?
Log In for instructions on accessing this content.
Pricing
Individuals
| AIChE Pro Members | $150.00 |
| AIChE Emeritus Members | $105.00 |
| AIChE Graduate Student Members | Free |
| AIChE Undergraduate Student Members | Free |
| AIChE Explorer Members | $225.00 |
| Non-Members | $225.00 |