(92c) Recovery of Valuable Metals from Industrial Leachates of Spent Ni Metal Hydride Batteries By Precipitation Processes.
AIChE Annual Meeting
2023
2023 AIChE Annual Meeting
Particle Technology Forum
Crystallization in Process Development
Thursday, November 9, 2023 - 8:50am to 9:15am
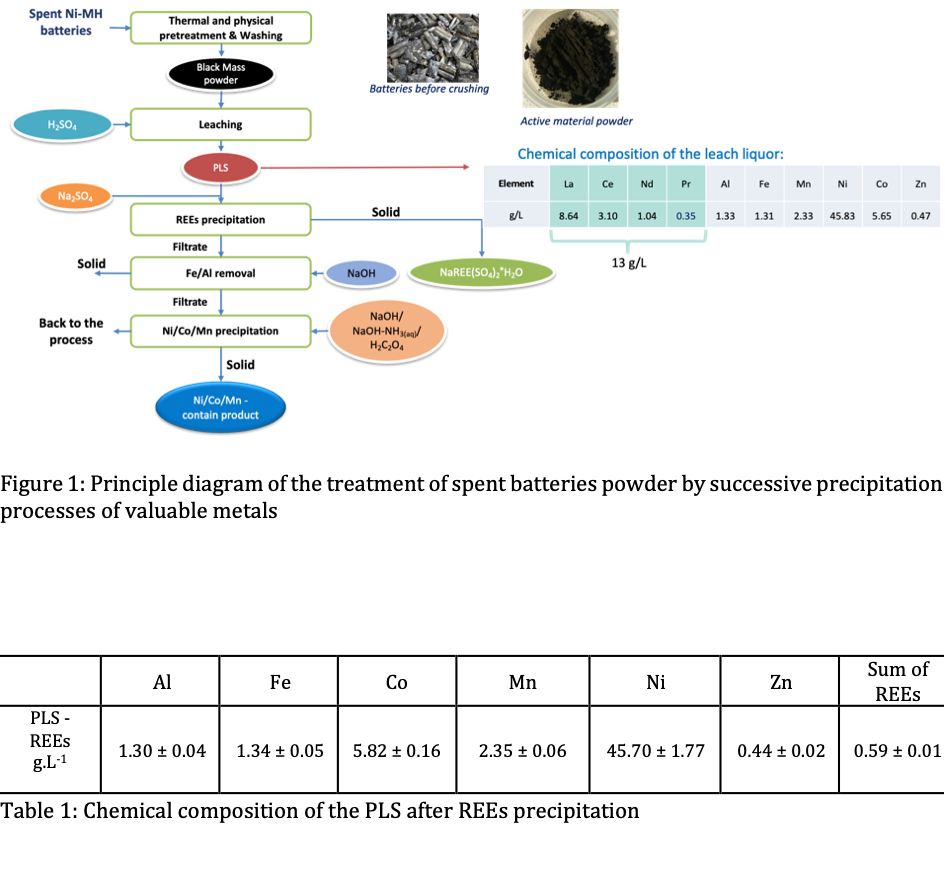
One possible flowsheet of selective separation of the critical metals by successive precipitation is proposed in Figure 1. The first step after leaching, often reported in the literature, is the recovery of rare earths elements (REEs) by precipitation from pregnant leach solutions (PLS) in sulfate media, using Na2SO4 [3]. However, little consideration is given as to whether and how sodium ions influence the precipitation efficiency and selectivity, and detailed phase characterization of the products is rarely reported. After REEs separation the obtained filtrate contains a high amount of Ni that can be recovered by specific precipitation. Nevertheless, before producing Ni components, it is necessary to withdraw impurities such as Fe and Al contained in the PLS by selected preliminary precipitation processes. Very few routes of Fe/Al elimination from the real leachates were explored in the literature. We propose to integrate this step in the recycling process. Finally, several possibilities of Ni precipitation from the final purified leachate can be envisaged.
This work focuses on a better understanding of the precipitation phenomenon and mass balances in the industrial PLS complex system and for the three successive precipitation steps (REEs, Fe/Al, Ni), by coupling pilot-scale experiments on industrially-sourced PLS, in-depth precipitated particles identification, and thermodynamic calculations.
The initial PLS used in this study was provided by SNAM company and originates from the pilot-scale leaching of industrial samples of spent Ni-MH battery powders in H2SO4 media, according to a process described in [2]. The battery waste leachate contains 46 g.L-1 Ni, 13 g.L-1 REEs and 11 g.L-1 of other metals like Co, Mn, Al and Fe.
We have applied a selective precipitation pathway to recover REEs and Ni from such a complex solution, without entrapping impurities such as Fe and Al. In our study, for each precipitation experiment, 1.5 L of PLS was added to a 2L double-jacketed agitated batch reactor (550rpm) and heated at 60°C; solution temperature was regulated and pH was monitored. In a standard experiment, a 2.9 M Na2SO4 solution was maintained at 40 °C and added to the PLS at a constant flow rate. Suspension volumes were regularly sampled, filtered, and solutions were analyzed by ICP-OES. The precipitation efficiency was calculated as the proportion of the REEs that have precipitated to the initially present in the solution. At the end of Na2SO4 addition, the suspension was kept stirred at constant temperature for 1 h and was then filtered to recover a Ni-rich solution on the one hand and REEs-rich filtered cake on the other hand. The obtained REEs crystals were washed and dried before in-depth characterization using multiple techniques: SEM-EDX, powder XRD, TGA, microwave digestion in aqua regia and ICP-OES.
An experimental parametric study on temperature, Na:REE ratio and Na2SO4 flowrate, allowed to obtain suited chemical conditions (Na:REEs = 3.6 and T = 60 °C), for high (>95%) REEs precipitation yields. We showed that the precipitation was selective i.e. only REEs precipitate while Ni and other elements remain in solution.
In parallel, the variation of REEs precipitation yield from the PLS via Na2SO4 addition was evaluated by thermodynamic modeling using the OLI studio module (v11.0) of the OLI.Systems software which includes an accurate thermodynamic description of rare earth sulfates recently developed by Anderko et al.[4]. This program implements the Mixed-Solvent Electrolyte (MSE) model for concentrated electrolyte solutions. The OLI-MSE database contains a specific thermodynamic description of the system rare earths sulfates/sodium sulfate/sulfuric acid /water and thus is well adapted to represent the chemical system.
Calculations were carried out with the various initial PLS conditions (pH, temperature, and ion concentrations). They foresee the precipitation of the compounds NaLa(SO4)2.H2O, NaCe(SO4)2.H2O, NaNd(SO4)2.H2O and NaPr(SO4)2.H2O in all conditions, which is in good agreement with XRD characterizations. The calculated REEs aqueous concentrations were also close to experimental measurements. At 60 °C, calculations show that the solubilities of all lanthanide-alkali double sulfates decrease with increasing Na+ concentration, which explains why increasing the Na:REEs molar ratio improves precipitation efficiencies at constant pH. Moreover, for a Na:REEs molar ratio of 4:1, increasing the temperature decreases the double sulfates solubilities (apart from NaCe(SO4)2.H2O which shows the opposite trend).
The XRD analysis of the product as well as of synthetic double sulfate salts allowed us to prove that the obtained product was a solid solution rather than a mixture of individual salts as it was supposed in previous works. The chemical formula of the obtained phase was (Na0.9K0.1) (La0.65Ce0.24Pr0.04Nd0.07)(SO4)2êžH2O, where the relative proportion of REEs was the same as in the initial PLS.
The second step of the treatment is the withdrawal of Fe and Al impurities, leaving the valuable elements Ni, Co, Mn in the PLS. The composition of the PLS after REEs precipitation is given in Table 1. The potential of the initial solution was measured and was about 0.64 V vs. SHE at 60 °C. Knowing that the potential of the solutions is driven by the couple Fe3+/Fe2+ in this system, and taking the Nernst equation, the ratio Fe2+/Fe3+ can be calculated and equals 20, which corresponds to 5% of Fe3+from Fetotal. Meanwhile, Fe3+ precipitates at pH < 3, while Fe2+ can be completely removed from the solution by precipitation only at pH around 8. The latter will affect the quality of the Ni-contain product, which can precipitate also in the same pH range. Hence, we propose to use H2O2 as an oxidizing agent in order to transform the portion of Fe2+ existing in the solution in Fe3+before precipitation with NaOH solution, allowing the removal of all iron at low pH. Then, pH was increased until pH=5 for precipitation of Al3+. The results show that accurate pH choice is a key factor for the selective recovery of Fe and Al from the PLS. The enhanced precipitation of Fe (>99%) was obtained via the oxidation of Fe2+ to Fe3+ using H2O2 solution. On the other hand, the obtained results also highlight the complete Al removal avoiding Ni losses. Thermodynamic calculation predicted precipitation of 99.9% of Al at pH 4.9.
The final step is to find a method for recovering Ni from the purified PLS. Several methods were investigated, leading to hydroxide or oxalate precipitation according to the selected reactant. The same precipitation efficiencies were reached using both methods: 98.1% of Ni precipitated at pH 9.5 as a hydroxide, while 99.6% of Ni precipitated at pH 0.2 in oxalate form. Nevertheless, in the case of hydroxide precipitation, an amorphous product with poor filterability (residual moisture of 77 wt%) was obtained which affected the washing and resulted in a high Na content in the solid phase. These observations were confirmed by XRD analysis.
In contrast, the precipitated oxalate had a well-crystallized structure and XRD analysis detected only one major phase, which corresponds to nickel oxalate dihydrate. This was confirmed by TGA analysis. The absence of the sodium sulfate on the diffractogram confirms the low Na content, determined by ICP-OES analysis
The study of the final stage of leach solution treatment showed the possibility of complete Ni, Co and Mn removal using either 4 M NaOH or 3.8 M H2C2O4 solutions. However, the obtained hydroxide had a high crystal structure disorder, regardless of the use of the seeding technique to prevent the formation of α–Ni(OH)2 polymorph, which led to a poor washing. As a result, the solid phase had high sodium content due to the retaining of the mother solution concentrated in Na+ and SO42-, and the particles were highly agglomerated. The precipitated oxalate NiC2O42H2O, in contrast, had a well-crystalized structure that allowed to eliminate the undesirable impurities from the solid phase.
[1] L. Cassayre, B. Guzhov, M. Zielinski, B. Biscans, Chemical processes for the recovery of valuable metals from spent Nickel Metal Hydride batteries: a review. Renewable and Sustainable Energy Reviews 170 (2022) 112983. doi:10.1016/j.rser.2022.112983
[2] M. Zielinski, L. Cassayre, P. Destrac, N. Coppey, G. Garin, B. Biscans, Leaching mechanisms of industrial powders of spent nickel metal hydride batteries in a pilot-scale reactor, ChemSusChem. 4 (2020) 616–628. doi:10.1002/cssc.201902640.
[3] A. Porvali, B.P. Wilson, M. Lundström, Lanthanide-alkali double sulfate precipitation from strong sulfuric acid NiMH battery waste leachate, Waste Manag. (2017). doi:10.1016/j.wasman.2017.10.031.
[4] G. Das, M.M. Lencka, A. Eslamimanesh, P. Wang, A. Anderko, R.E. Riman, A. Navrotsky, Rare earth sulfates in aqueous systems : Thermodynamic modeling of binary and multicomponent systems over wide concentration and temperature ranges, J. Chem. Thermodyn. 131 (2019) 49–79. doi:10.1016/j.jct.2018.10.020.