(696a) Predicting Metal Dynamics, Surface, and Segregation Energies in High Entropy Alloys for Sintering and Catalyst Durability
AIChE Annual Meeting
2023
2023 AIChE Annual Meeting
Catalysis and Reaction Engineering Division
New Developments in Computational Catalysis II: Data-Driven Methods
Tuesday, November 7, 2023 - 12:30pm to 12:50pm
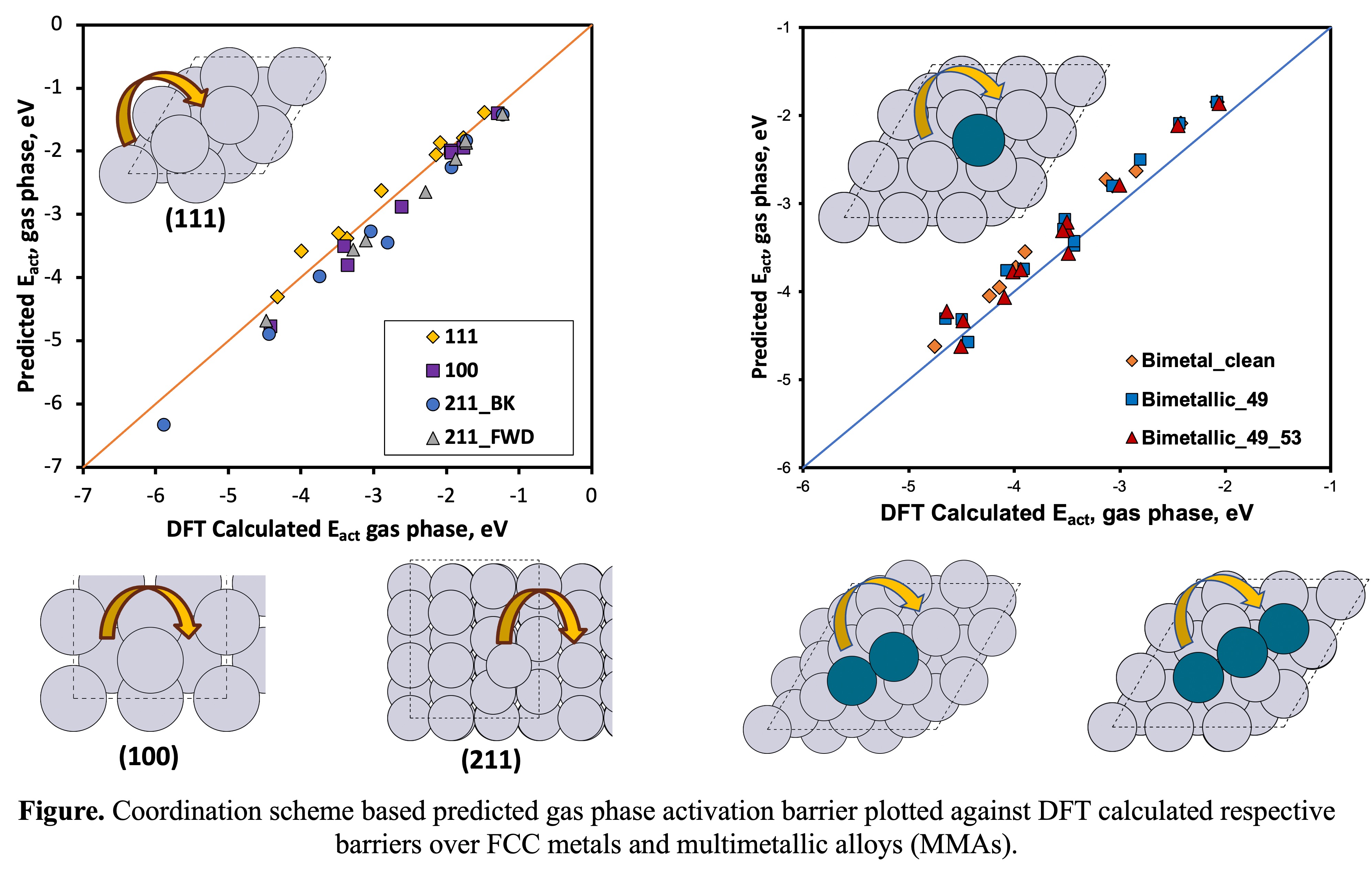
Moreover, the optimization of catalytic activity and selectivity in heterogeneous catalysis relies on various surface properties, particularly surface energies, and the distribution of site morphologies. Therefore, we also evaluate surface energies and predict the relative stabilities of various catalytic surfaces using the mentioned coordination schemes. We use periodic slabs and various metal nanoparticle (MNP) shapes to determine surface energies and extended this approach to multimetallic surfaces (alloys) to describe segregation behaviors in multimetallic alloys (MMAs) to predict the relative surface concentrations of the various constituent metals.
Overall, this physics-based approach allows for the effective screening of thermodynamic stabilities of alloy MNPs by integrating coordination schemes with structural and site information. The resulting models can be applied to calculate the energetics of any nanoparticle morphology and chemical composition, thus significantly accelerating the design of durable nanoalloys, potentially extending to high entropy alloys.
1. Roling, L.T., The Journal of Physical Chemistry C, 2017. 121(41): p. 23002-23010.