(627c) Entropic Analysis of Antigen-Specific CDR3 Domains Identifies Essential Binding Motifs Shared By CDR3s with Different Antigen Specificities
AIChE Annual Meeting
2023
2023 AIChE Annual Meeting
Food, Pharmaceutical & Bioengineering Division
Systems and Quantitative Biology: Disease Mechanisms, Biomarkers, and Therapies II
Monday, November 6, 2023 - 8:36am to 8:54am
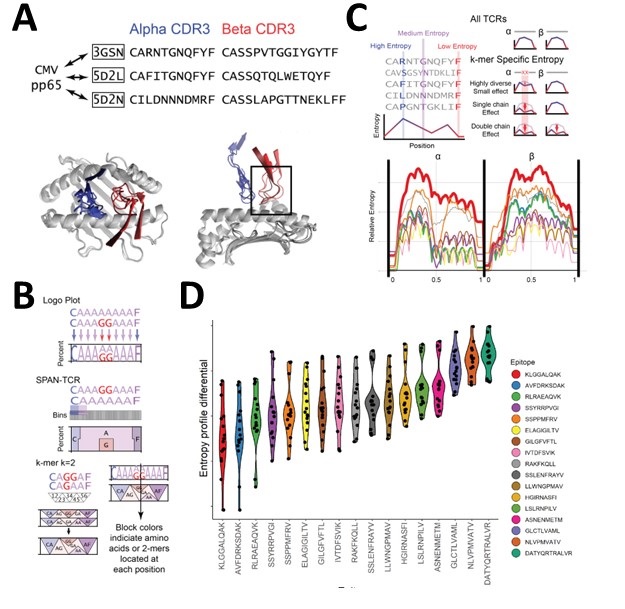
Materials and Methods: Here we present Scanning PArametrized by Normalized TCR Length (SPAN-TCR) as a tool for extracting structural and chemical insights from groups of antigen-specific TCR sequences in a length-agnostic fashion. We use SPAN-TCR to first describe the relative positions of amino acids and amino acid k-mers in CDR3 chains. We postulate that if an amino acid 2-mer (YZ) is important for binding to a specific pMHC, then YZ in XYZX is likely performing a similar function to YZ in XXYZXX. We then use SPAN-TCR to calculate informational entropy to identify high frequency 2-mers that we label as ‘essential’ or ‘super-essential’. An essential 2-mer is one that lowers the informational entropy (sequence diversity) within its own (α or β) CDR3 chain, while a super-essential 2-mer lowers the informational entropy within both CDR3 chains. We hypothesize that such 2-mers are important for TCR-pMHC binding, and we test this hypothesis by probing for 2-mer interfacial chemical interactions using molecular dynamics simulations. Finally, we extend these SPAN-TCR algorithms to yield comparisons between sets of TCRs known to bind to different antigens. SPAN-TCR is first validated through the analysis of public data bases of TCRs specific to viral antigens, followed by explorations of newly sequenced putative antigen-specific CDR3s against COVID-19.
Results and Discussion: We first analyze sets of TCRs specific to common viral antigens, finding similarities between SPAN-TCR and other methods such as GLIPH. We also use the SPAN-TCR framework to confirm that CMV-targeted pull-down of T cells from a human sample are likely antigen-specific by comparison to known TCRs. Entropic analysis is used to find entropy-reducing essential and super-essential k-mers in CMV-specific TCRs, and the location and identity of essential and super-essential k-mers within TCRs reveals trends, showing that hydrophilic and charged amino acids are the most consistently entropy-reducing, despite being uncommon within TCRs. These trends are confirmed in a large COVID-19 data set of putative antigen-specific TCRs, and validated in silico using Alphafold simulations. Finally, we find that patterns of entropy-reducing k-mers within sets of TCRs appear to be correlated to the amino acid sequence of the antigen itself.
Conclusions: SPAN-TCR is a suite of tools for the analysis of large databases of diverse TCR CDR3 sequences. It represents a shift away from the sequence alignment school of thought when comparing biological sequences. It is unique among analytical tools for TCR analysis in the way it is able to compare TCRs of different lengths to distill critical information for antigen-specificity in the form of essential entropy reducing k-mers. These k-mers can be pursued as anchor points for TCR design and engineering.
Figure caption: SPAN-TCR analysis of TCRs. A. TCRs have been observed with regular binding patterns when targeting the same antigen. These patterns lead us to model TCR binding with different methods than sequence matching, at less computational cost than molecular dynamics, even for TCRs of different lengths. B. A Logo plot is often used to describe a set of biological sequences by the frequency of amino acids used. We develop a modified version of the Logo plot that considers sequences of different lengths and models the contribution of consecutive amino acids (k-mers). C. Entropic analysis identifies k-mers that, when found in a TCR, drastically reduce the entropy of a set of TCRs in a single chain, both chains, or not at all. These k-mers that reduce entropy are likely essential to TCR binding. D. Patterns of entropy-reducing k-mer usage inside the TCR shows antigen target-specific patterns.