(75f) Towards Miktoarm Polymers Via Sequence-Defined Oligocarbamate (SeDOC) Initiators
AIChE Annual Meeting
2022
2022 Annual Meeting
Materials Engineering and Sciences Division
Polymer Synthesis and Reaction Engineering
Monday, November 14, 2022 - 9:30am to 9:45am
Star block copolymers with branched architectures ((AB)n) have often been shown to have higher tensile strength than their linear counterparts, but also tend to have a lower elastic recovery. The core’s star architecture more efficiently distributes stresses, leading to upgraded mechanical properties. Furthermore, branched block copolymers have been shown to display thermodynamic properties that cannot be attained by their current linear counterparts. Thus, the controlled synthesis of block copolymers with branched architectures provides a unique opportunity to discover new phases beyond traditional linear block copolymer phase behavior.
Miktoarm star polymers, which have heterogeneous arms connected at a common core, have been shown to reduce domain spacing and stabilize new and interesting phase behaviors. However, the synthesis of precise asymmetric miktoarm polymers is non-trivial and requires extensive synthetic manipulation. While the synthesis of symmetric star polymers can be achieved using dendrimer and other synthetic scaffolds, the introduction of the asymmetric arm is much more difficult and can only be achieved through tedious synthetic routes and inefficient polymeric coupling reactions. Modulating the number of arms and making precise cores with multiple asymmetric arms is laborious to accomplish at scale. This can be addressed by leveraging a recently developed sequence-defined oligocarbamate (SeDOC) platform to synthesize precise multivalent initiators that will lead to the assembly of symmetric star polymers and asymmetric miktoarm polymers. These precise multivalent initiators were used for the controlled ring-opening transesterification polymerization (ROTEP) of γ-methyl-ε-caprolactone (γMCL) and L-lactide (LLA) that will lead to the assembly of star polymers with rubbery inner blocks and semi crystalline and glassy hard blocks.
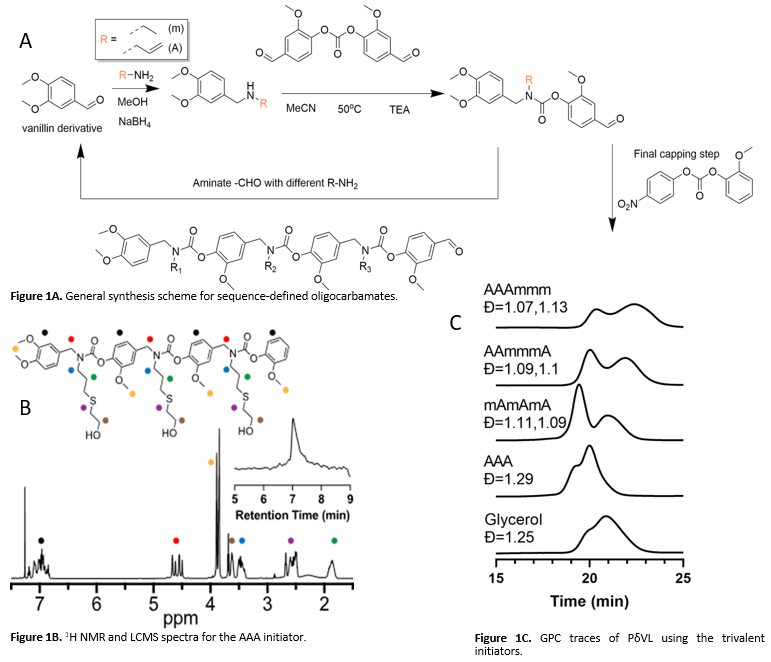
The synthesis of SeDOCs can be achieved using a sustainable feedstock, vanillin, and high-yielding reductive amination and carbamate formation reactions (Figure 1A). The SeDOCs have modifiable pendant groups and a rigid aromatic vanillin-based backbone. The reactive pendant groups of the SeDOCs are then appended with β-mercaptoethanol to give hydroxyl-terminated initiators.
Previous work done in the Alabi research group shows the importance of monomer sequence on the properties of cross-linked networks. This inspired the current investigation into the effect of initiator core spacing on the resulting polymerization efficiency. In this study, we show that the SeDOC platform can be used to control how polymers are distributed across the arms of an initiator by varying the initiator sequence. This work utilizes a green chemical platform for the preparation of star polymers. The core and polymer arm segments are degradable and created from biorenewable sources. A library of initiators was synthesized, and each initiator is named according to its pendant group sequence, where “A†represents a reactive pendant group that can be initiated and “m†represents an unreactive pendant group that does not participate in initiation. The library included a monovalent initiator (mAm), a divalent initiator (AmA) and several trivalent initiators with varying core spacing (AAA, AAAmmm, AAmmmA and mAmAmA). SeDOC initiators were characterized using 1H NMR and liquid chromatography mass spectroscopy (LCMS) (Figure 1B). Control experiments were carried out with commercially available with monovalent (benzyl alcohol, BA), divalent (1,4-benzenedimethanol, BdM) and trivalent (glycerol) initiators to compare with the polymerizations done using SeDOC initiators.
To understand the effect of core spacing on ROTEP efficiency, experiments were performed in bulk using δ-valerolactone (δVL) as the monomer and diphenyl phosphate (DPP) as the catalyst. The polymers were synthesized and characterized via 1H NMR and gel permeation chromatography (GPC). The mAm and AmA initiated polymers had similar molecular weight distributions and polydispersity values to their corresponding control compounds in BA and BdM. While the molecular weight distributions and dispersity of the polymers made using AAA and glycerol were similar, they were different from the polymers made using the longer trivalent initiators AAAmmm, AAmmmA and mAmAmA (Figure 1C). The longer sequences had a distinct bimodal distribution when compared with the AAA initiator and was most pronounced in the mAmAmA initiator. The bulk polymerization kinetics of these initiators were monitored via 1H NMR and it was observed that mAmAmA initiator reaches a significantly lower conversion percentage than the other trivalent initiators. Similar trends was also observed in the solution phase kinetics. While monitoring the kinetics through GPC, the bimodal distribution of the mAmAmA initiator persists through the duration of the polymerization while the AAA initiator starts with a bimodal distribution that coalesces into a single peak during the course of the experiment. These experiments have shown that the resulting polymer distribution is influenced by the sequence-specific nature of the initiators. To disentangle the effects due to monomer crystallinity and initiator kinetics, the subsequent experiments were done using an amorphous monomer, γ-methyl-ε-caprolactone (γMCL).
Further ROTEP experiments were done in bulk using γ-methyl-ε-caprolactone (γMCL) as the monomer and Sn(Oct)2 as the catalyst. This was followed by the orthogonal ROTEP of L-lactide in dichloromethane using 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as the catalyst resulting in (PγMCL)-b-(PLLA) star block polymers. Growth of PLLA from the γMCL-SeDOC core is confirmed by 1H NMR and a clear increase in the molar mass of the (PγMCL)-b-(PLLA) polymers from their parent (PγMCL)n core is observed via GPC. As the number of arms in the initiator increases (from mAm to AAA), the volume fraction of PLLA (fPLLA) in the polymer decreases. It was also observed that fPLLA and overall molar mass varied with the composition and sequence of the trivalent SeDOC initiators. During the ROTEP of γMCL, the initiators without the spacing between the arms, i.e. AAA and AAAmmm, form polymers with higher Mn than the initiators with spacers. Similar experiments were also carried out using A4, A5 and A6 initiators and were compared with the AAA initiator. The A4 and A5 initiators were similarly effective and had comparable fPLLA values as the AAA initiator. However, there was a steep decline in the effectiveness of polymerization that was observed in the case of the A6 initiator, which was due to the transesterification of the polymeric arms leading to shorter chains than expected. Overall, our results show that changes in the sequence of the initiator affects the resulting polymer molecular weight distribution and dispersity. This response in molecular weight distribution to the subtle change in initiator sequence could be used to tune the properties of the elastomers through the straightforward implementation of a star architecture.
The next steps include characterizing the mechanical and thermal properties of the polymers that have been made to determine how these properties are affected by molecular weight distribution. We have demonstrated the efficient synthesis of a series of aliphatic polyester star block TPEs using the SeDOC initiators and can target different architectures using the sequence-specific nature of the initiator. Comparison to commercially available analogues indicates that the change molecular weight distribution is a feature of the star architecture. This work has shown that we are able to tune the resulting distribution of TPEs through the straightforward implementation of a star architecture. The enhanced understanding of the relationship between the polymer architecture and material properties from the future work will allow for improvements in the design of high-performance sustainable TPEs.