(478d) Examination of a Hybrid Catalytic System for Electrochemical Reduction of CO2
AIChE Annual Meeting
2022
2022 Annual Meeting
Catalysis and Reaction Engineering Division
Electrocatalysis II: Catalyst and Characterization
Wednesday, November 16, 2022 - 1:24pm to 1:42pm
All DFT energies were computed using the VASP code with the VASPsol extension for considering solvation effects. In addition to implicit solvation, explicit water molecules were also included to account for strong hydrogen bonding where appropriate.
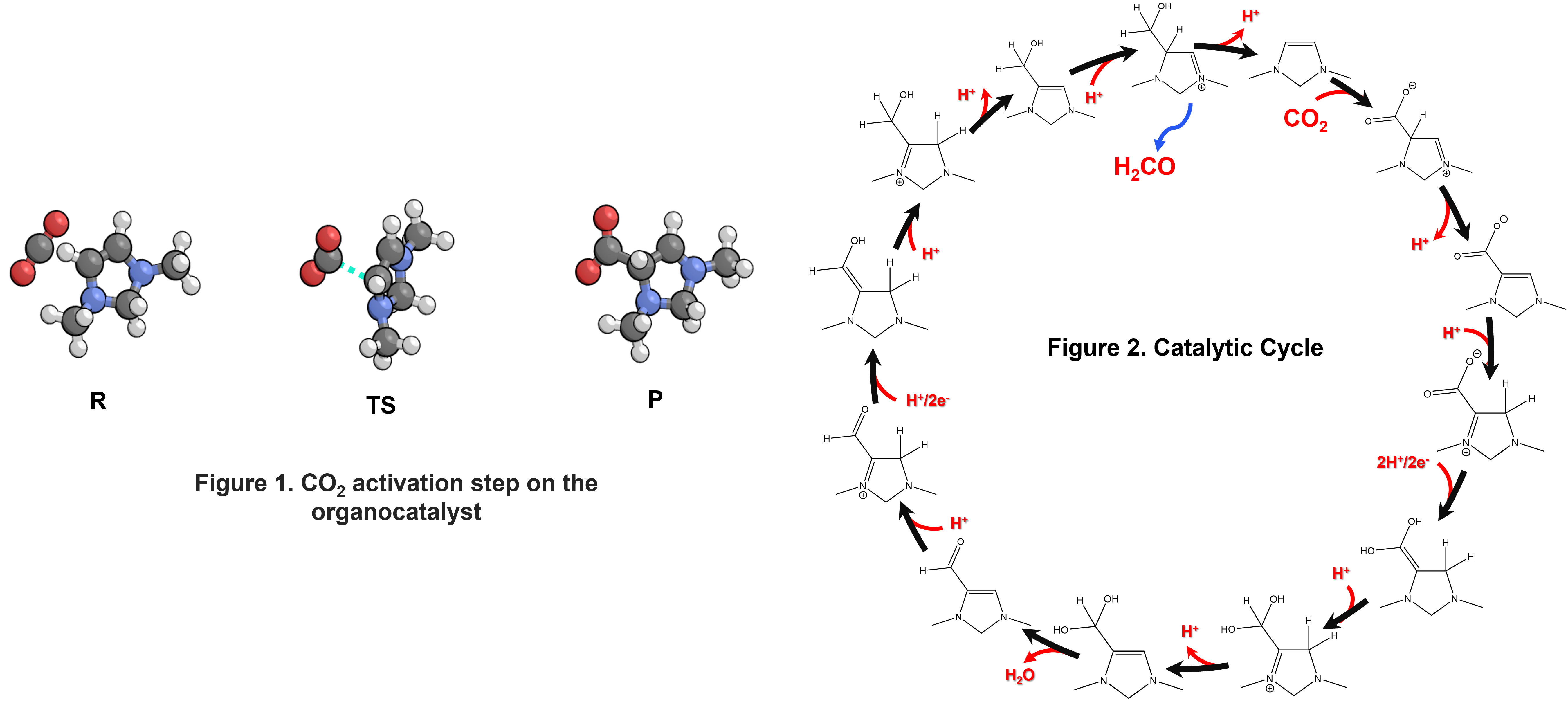
The organocatalyst is a reduced 1,3-dimethylimidazolium whose formation is predicted to be thermodynamically favorable at reaction conditions. This species is found to readily undergo CO2 addition (Figure 1) at the C-4 carbon, with a transition state free energy that is 0.57 eV above the resting state. The addition product undergoes a series of tautomerization and proton-coupled electron transfer steps (Figure 2) before eliminating formaldehyde to complete the catalytic cycle. At the optimal pH of 4.1 and potential of -0.6 V vs. RHE, the reaction is calculated to have a low effective activation free energy barrier of 0.73 eV. These low barriers are contingent upon having an effective metal-sulfide-based proton transfer catalyst with a pKa equal to the pH. Although the metal sulfide surface was not explicitly included in the calculations, its effect was approximated by using a series of sulfur-based proton donors to construct a linear relationship between the sulfide pKa and the activation barrier. Future work will focus on identifying such a suitable metal sulfide surface and explicitly including it in the DFT calculations.