2022 Annual Meeting
(619b) Theoretical Modeling of Interfacial Proton-Coupled Electron Transfer
Authors
Robert Warburton - Presenter, Yale University
Sharon Hammes-Schiffer, Yale University
Alexander Soudackov, Yale University
Phillips Hutchison, Yale University
James M. Mayer, Yale University
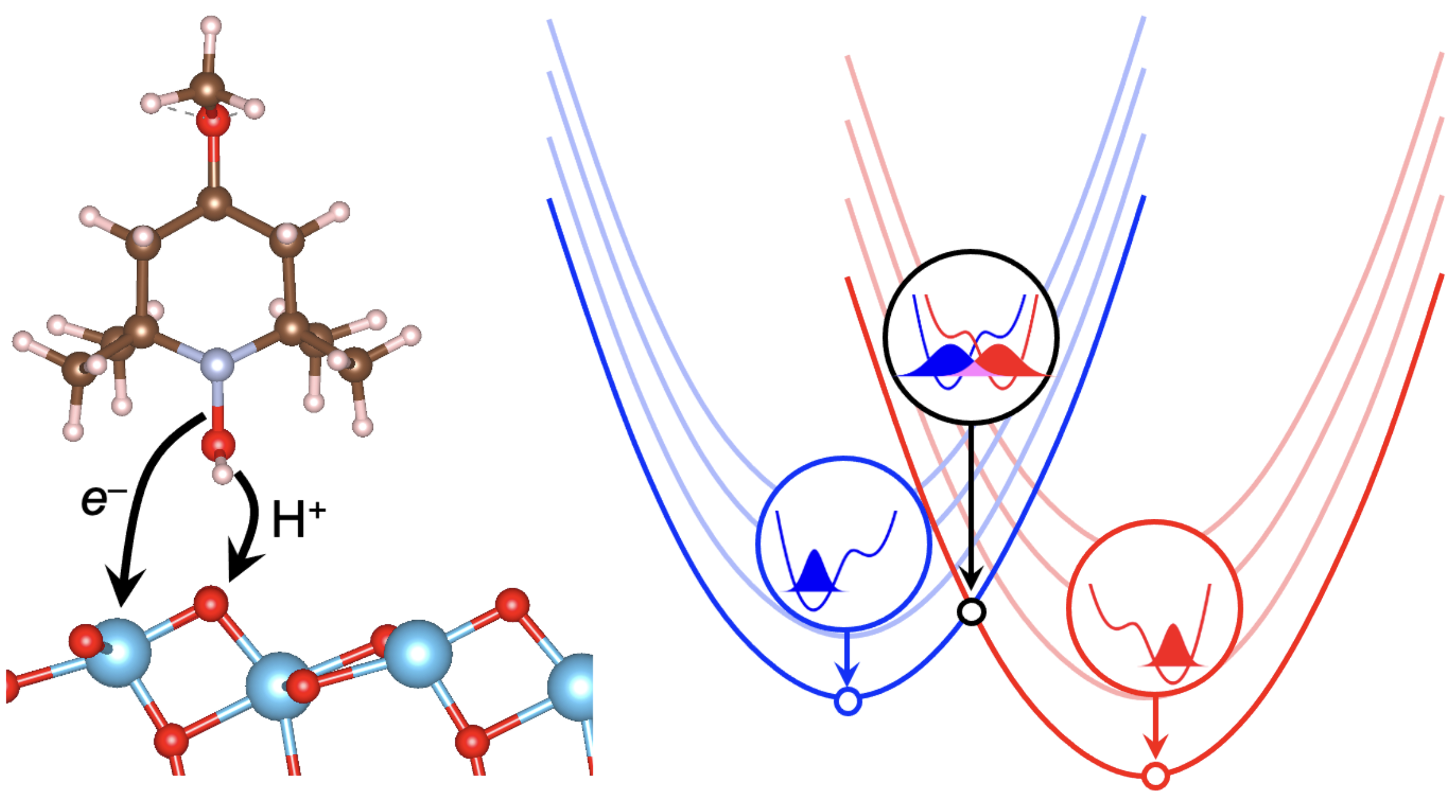
Proton-coupled electron transfer (PCET) reactions are elementary steps in biological processes such as respiration, as well as in many electrocatalytic transformations. Given the fundamental role of PCET in the mechanisms of energy-critical catalytic reactions, it is important to understand the underlying physics of this vital class of redox reactions at interfaces. Here, periodic density functional theory (DFT) is used to model the thermodynamics and kinetics of interfacial PCET using two well-defined systems. We first investigate electrochemical PCET at graphite-conjugated catalysts, where organic acids conjugated to solid carbon surfaces undergo heterogeneous PCET with molecular tunability. The impact of interfacial electrostatic potentials with applied potential bias and the importance of continuous conjugation for heterogenous PCET is demonstrated. Proton-coupled redox potentials calculated using both constant charge and constant potential methods are in close agreement with experimental results. Next, hybrid functional DFT calculations are used to analyze PCET at anatase TiO2 surfaces and their relationship to electronic defects within the TiO2 band gap. Calculated OâH bond dissociation free energies for geometrically similar bonds vary by ~81 kcal/mol (~3.50 eV) depending on whether charge-compensating carrier defects are conduction d-band electrons or valence p-band holes. These differences are shown to be primarily due to ET driving forces, analyzed within a Marcus theory framework as a combination of band energies and inner-sphere reorganization energies. The kinetic reactivity of different trap states with a nitroxyl radical PCET oxidant is analyzed using vibronically nonadiabatic PCET rate theory, including the contributions of excited vibrational states to the overall rate constant. These computational studies are compared to experimental optical titrations of electrons in TiO2 trap states. These theoretical studies describe the interplay between ion and electron reactivity at interfaces.