2022 Annual Meeting
(610i) Prediction of Metal-Organic Interactions and Molecular Assembly in High Accuracy and Speed
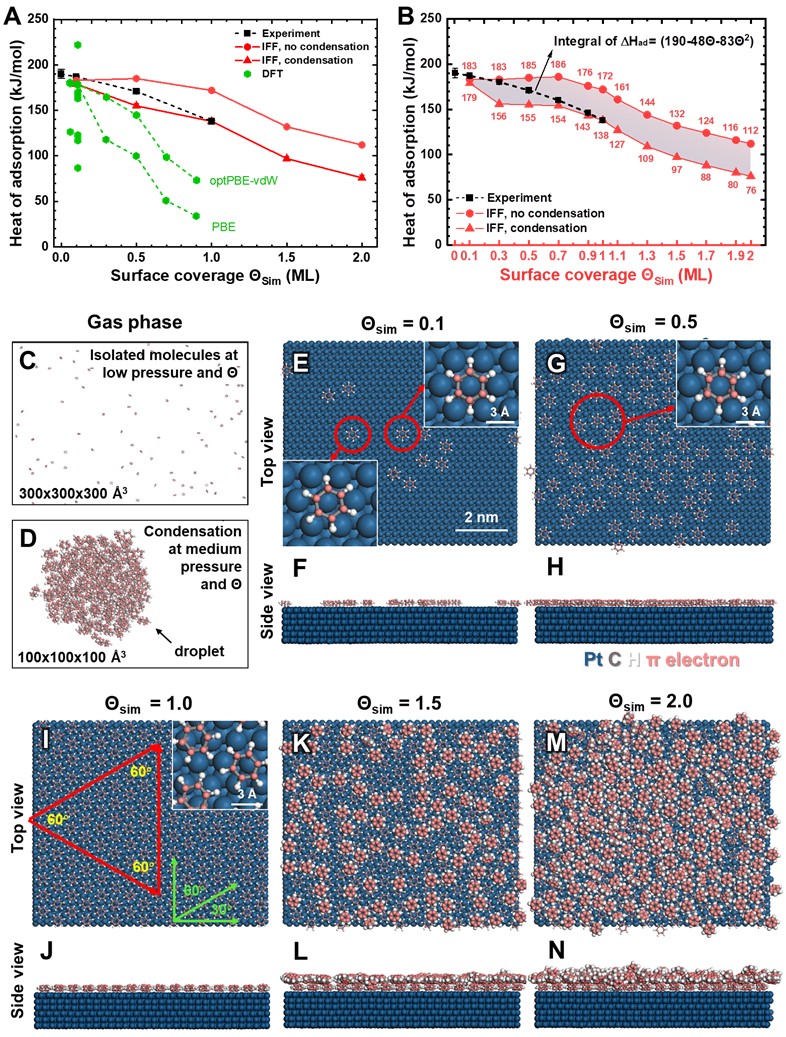
Understanding and predicting metal-organic interactions at the atomic scale is critical for the development of materials for energy conversion, sensors, and therapeutics. Major bottlenecks to-date include scarce experimental data, and lack of facile methods to reliably and fast predict important binding and assembly preferences at the atomic scale. Here we show predictions of adsorption energies of aromatic molecules on platinum surfaces in unrivalled accuracy by molecular dynamics simulations with the Interface Force Field (IFF) in comparison to extensive experimental measurements in highest quality. The accuracy is up to 15 times higher than with density functional theory at 1 million times faster computational speed and allow 2 to 4 times better binding predictions relative to CHARMM-IFF. Quantitative insights into molecular recognition, assembly, the role of various defects, and condensation in the gas phase are described. We demonstrate that the methods are applicable to trillions of unknown metal-organic and metal-electrolyte interfaces for accurate and efficient screening.