2022 Annual Meeting
(572a) Active Learning for Efficient Navigation of Adsorption Landscapes in MOFs
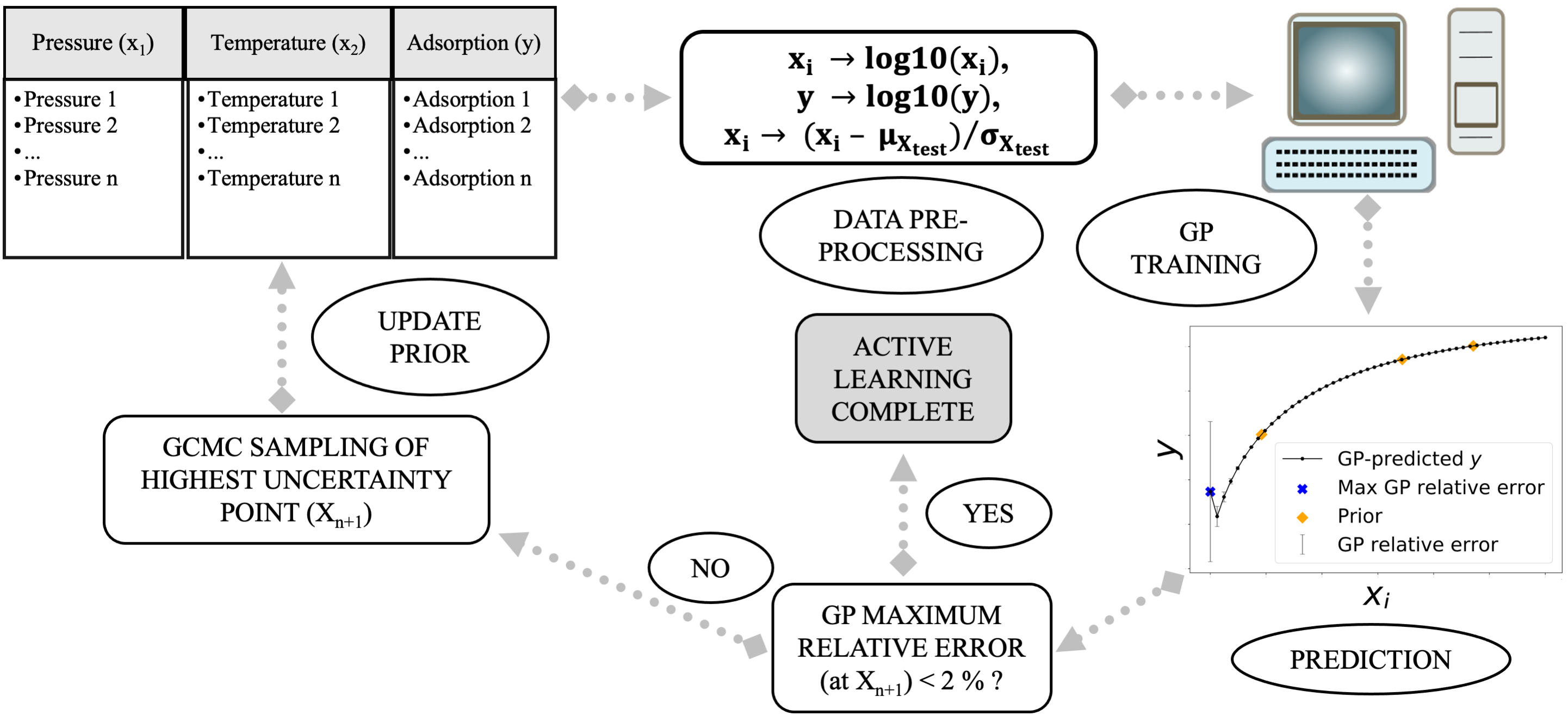
Machine learning (ML)-based surrogate models are becoming popular in applied sciences since they are computationally inexpensive and have great predictive ability.1 Also, these cheap ML-based models can replace the conventional simulation methods for future predictions. However, one needs a large set of training data to build a ML-based surrogate model and often these datasets are expensive to generate. This is particularly true for adsorption where data generation is costly as they require molecular simulations which can take a few minutes to a few days to finish. This creates a bottleneck in scaling these models to add multiple features. In this prospect, we present an active learning (AL)-based protocol to predict gas adsorption in metal-organic frameworks (MOFs). AL builds the training set based on the uncertainty in its output prediction and thus it needs a smaller number of simulations to build itself.2 We fit this AL model sequentially, starting from a prior dataset created using grand canonical monte carlo (GCMC) simulations and then sequentially build this model by sampling the next posterior point using the uncertainty in the predicted adsorption on the test set. Gaussian process regression (GPR) is used as a regression model for emulating the GCMC simulations. We continue to build the AL model until the uncertainty in adsorption for each point on the test set is lower than a threshold value (which is decided a priori). In this work, we first show how AL can be employed to build surrogate models for pure methane and carbon dioxide adsorption isotherms; pressure is the sole feature. Finally, we showcase how a GPR model can predict adsorption landscapes (varying both pressure and temperature) with an order of magnitude smaller number of simulations. In this process, we also compare different prior selection criteria and evaluate their performance. Based on our results, we believe this AL-based protocol has potential for adsorption prediction in more complex systems.
References
[1] A. Sturluson, M. T. Huynh, A. R. Kaija, C. Laird, S. Yoon, F. Hou, Z. Feng, C. E. Wilmer, Y. J. Colón, Y. G. Chung, D. W. Siderius and C. M. Simon, Molecular Simulation. 45 (2019) 1082â1121.
[2] E. Uteva, R. S. Graham, R. D. Wilkinson and R. J. Wheatley, The Journal of Chemical Physics. 149 (2018) 174114.