2022 Annual Meeting
(41e) Spectroscopic Probe Molecule Selection Using Quantum Theory, First-Principles Calculations, and Machine Learning
Authors
Joshua Lansford - Presenter, University of Delaware
Dionisios Vlachos, University of Delaware - Catalysis Center For Ener
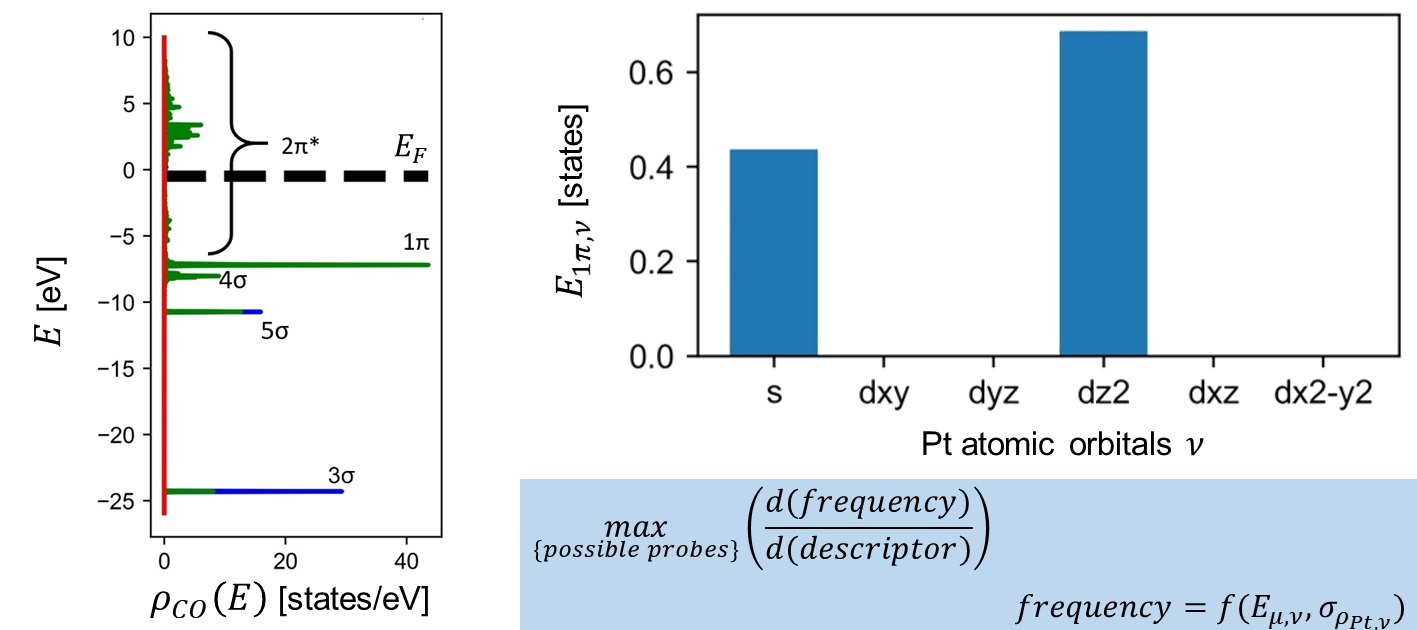
Probe molecule vibrational spectra have a long history of being used to characterize materials including metals, oxides, metal organic frameworks, and even human proteins. Furthermore, recent advances in machine learning have enabled computationally generated spectra to aid in detailed characterization of complex surfaces with probe molecules. Despite widespread use of probe molecules, the science of probe molecule selection is underdeveloped. Here, we develop physical concepts, including orbital interaction energy and the energy overlap integral, to explain and predict the ability of probe molecules to discriminate structural descriptors. We resolve the crystal orbital overlap population (COOP) to specific molecular orbitals and quantify their bonding character, which directly influences vibrational frequencies. Using only a single adsorbate calculation from density function theory (DFT), we compute the interaction energy of individual adsorbate molecular orbitals with adsorption site atomic orbitals across many different sites. Combining the molecular orbital resolved COOP and changes in orbital interaction energy enables probe molecule selection for improved discrimination of various sites. We demonstrate these concepts by comparing the predicted effectiveness of carbon monoxide (CO), nitric oxide (NO), and ethylene (C2H4) to probe Pt adsorption sites. Finally, using a previously developed machine learning framework, we show that models trained on hundreds of thousand C2H4 spectra, computed from DFT, which regress surface binding-type and generalized coordination number (GCN), outperform those trained using CO and NO spectra. A python package, pDOS_overlap, for implementing the electron density based analysis on any combination of adsorbates and materials, is also made available.