2022 Annual Meeting
(268f) Properties of Tri-Butyl-Phosphate from Polarizable Force Field MD Simulations
Authors
Over the years, several force fields (FF) have been developed (polarizable and non-polarizable) to predict the properties
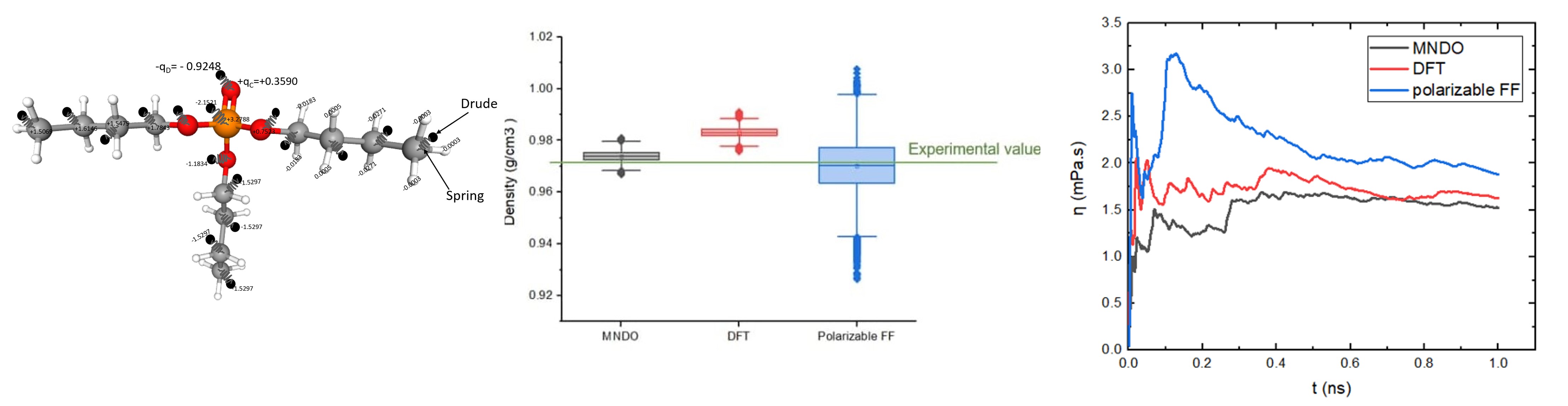
of TBP, and a number of articles have been published using non-polarizable classical molecular dynamics for studying various aspects of chemical complexation in solvent extraction containing TBP. However, the current capability of atomistic simulation to predict properties are relatively poor. In our previous study, the performance of the OPLS polarizable FF and two sets of non-polarizable force field models, namely OPLS-DFT and OPLS-MNDO for TBP in the liquid state have been examined to calculate properties of pure TBP. Following our previous work, we have obtained excellent results for predicting the mass density of TBP (as expected), for example, the mean value obtained is 0.97023 g/cm3 (sigma=0.011) at ambient conditions which gives 0.08% error when compared to the experimental value.
But we find that our MD simulations using both types of force fields cannot satisfactorily reproduce transport properties such as viscosity and self-diffusion coefficient, specifically, results tend to under-estimate these properties; some of the current results such as mass density and dynamic viscosity are shown in the following figures.
As these results may not be appropriate for reproducing the properties of TBP, further work is needed to explore the possibility of performing improved calibrated polarizable FF. We continue to investigate the possibility of using MD to compute properties with results closer to experimental values. MD simulations on liquid TBP were performed on systems in cubic cells and equilibrated in an isobaric-isothermal (N-P-T) ensemble at 1 bar and 298.15 K for 2 ns. We have been using the Drude package in the LAMMPS MD code to implement atomic polarization. In this work, we are specifically studying the performance of OPLS polarizable FF for TBP to compute: mass density, heat of vaporization, dynamic viscosity, self-diffusion coefficient, thermal expansion coefficient, and dipole moment. This investigation also constitute a comparison between polarizable and non-polarizable FF models, where TBP has both polar and non-polar groups of atoms. It is expected that this work can provide recommendations on force fields for TBP to be used in future additional studies.