2020 Virtual AIChE Annual Meeting
(496f) Molecular Dynamics Simulation Based Approach to Study Cryoconcentration of Solutes during Freezing and Its Impact on Protein Stability
Authors
Bulk protein therapeutics are generally frozen for long term storage to prevent product degradation and facilitate product transportation. The freezing process of bulk protein solutions leads to cryoconcentration of solutes, which may impact protein stability. However, the underlying mechanism and molecular details of proteins unfolding and aggregating at the cryoconcentration microenvironment are poorly understood. Molecular dynamics (MD) simulations have been developed to investigate how cryoconcentration of different solutes affects protein stability during the bulk freezing process. The simulations results from this current work have been validated with previous experimental data on solutes cryoconcentration acquired by our group.
Methods:
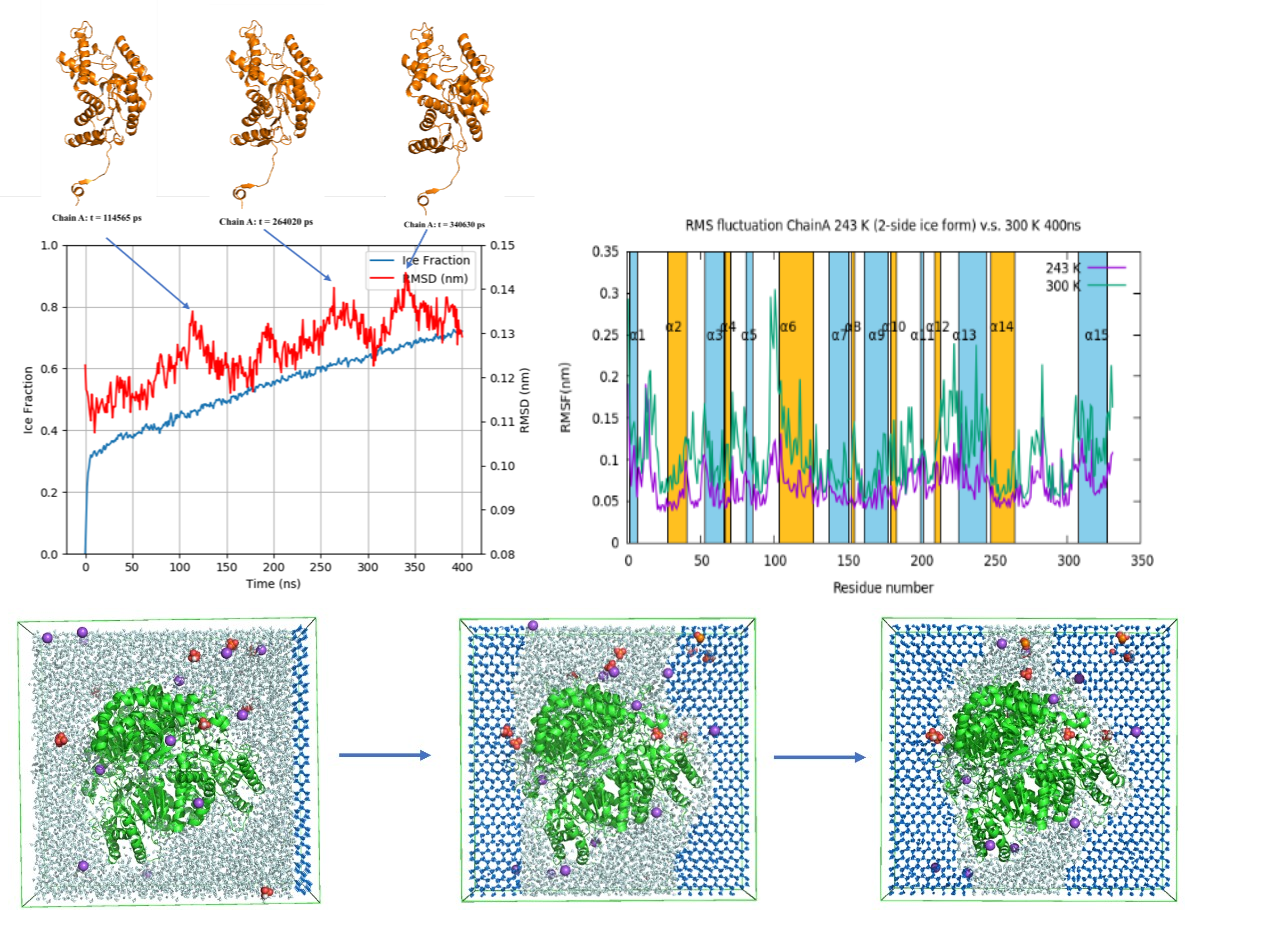
The initial conditions applied into the MD simulations were selected based on previous cryoconcentration experiments in which 1 L bottles containing 600 mL of a protein solution of 0.5 mg/mL lactate dehydrogenase (LDH) in 10 mM phosphate buffer at pH 7.0 were frozen at -30°C. Therefore, LDH and sodium phosphate saltsâ molecules were added into the simulation box. The MD simulation trajectories and their analyses were carried out using GROMACS, VMD and Pymol software. We applied both CHARMM and MARTINI force-field for all-atom as well as coarse-grained simulations, respectively. Moreover, Steered MD simulation has been applied to investigate potential mean force of protein stability related association and dissociation process. The initial coordinates for LDH were taken from the protein data bank entries (pdb: 5NQB). The protein was initially modeled using the psfgen tool in VMD, and then further solvated in a simulation box with TIP4P ice water model and charge neutralized with NaPO4. The box volume was optimized in the NPT ensemble by initially applying 500 cycles of a conjugate-gradient minimization scheme followed by a short 40-ps MD run in NPT assemble. The temperature was controlled using the Langevin thermostat, and the pressure was controlled by the Nose-Hoover barostat. The simulations were carried out using periodic boundary conditions. These briefly equilibrated systems of LDH protein were further subjected to MD simulation sets in the NVT ensemble. Computations were performed on HPC, a supercomputer cluster at the University of Connecticut.
Results:
The cryoconcentration effect was evaluated applying a model protein solvated in a simulation box of aqueous solvent in which different ions and stabilizers were added for case studies. Analyses of the resulted trajectories reveal the effect of temperature, ionic strength, and solutes concentration on the protein unfolding state, biomolecular interactions, and protein aggregation. The final concentration validation show a good match of simulation results and the previous experiment data.
Conclusions:
In this present work, we have identified the relatively more conformation variations of LDH at higher temperature which leads to unstable LDH condition, whereas, the stability has been reinforced with ice formation at lower temperature. In addition, the most flexible regions on protein corresponding with the most impact residues from ice-water interface were discovered successfully. To compare two types of ice formation cases, both-side ice formation revealed relatively more stable LDH protein conformation with less solvent accessible surface area and more hydrogen bonds. LDH protein aggregation has been identified at CG-MD at higher temperature and we discovered the possible residues aggregated based on statistics probability repeats.