2020 Virtual AIChE Annual Meeting
(477i) Physical Graph Neural Networks for Prediction of Fuel Ignition Quality
Authors
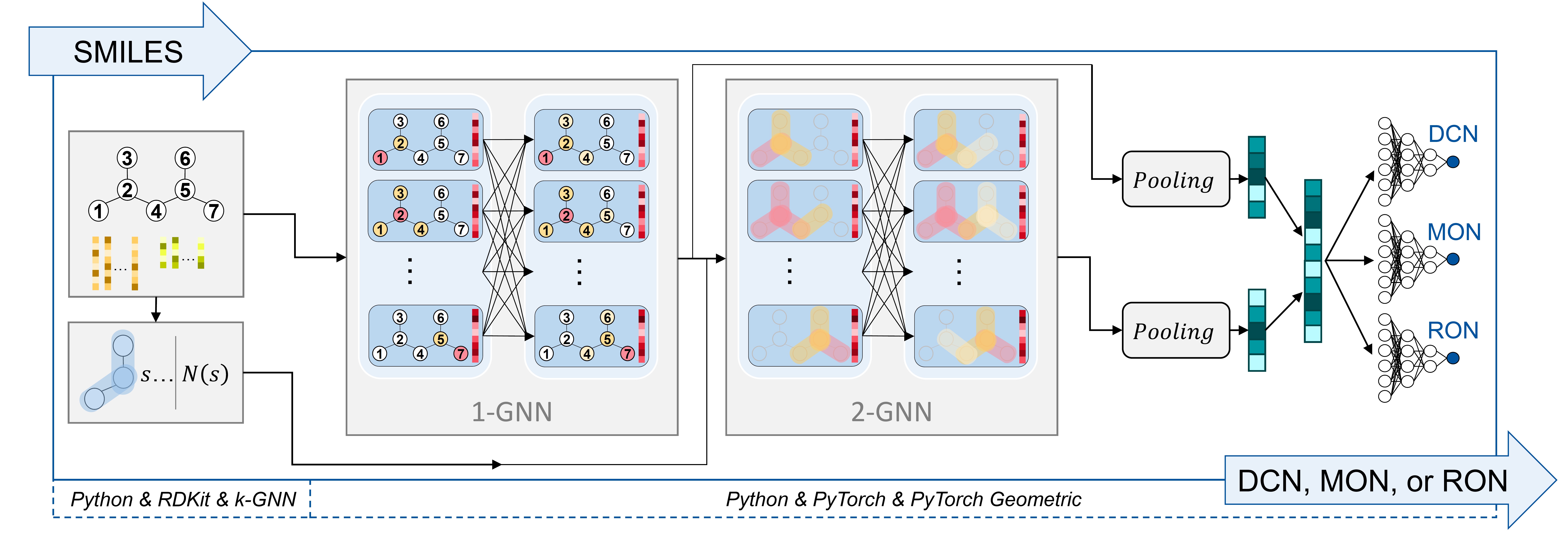
Recently, graph neural networks (GNNs) [6,7] have shown promising results for the prediction of structure-property relationships of molecules [8]. GNNs use graph representations of molecules, where nodes correspond to atoms and edges to bonds. These graph representations are then linked to molecular properties by means of graph convolutions, pooling layers, and multilayer perceptron networks. More specifically, GNNs learn physico-chemical properties as a function of the molecular graph in a supervised learning set-up using a backpropagation algorithm. This end-to-end learning eliminates the need for selection of functional groups or descriptors. Rather, the method learns optimal molecular fingerprints through graph convolutions and maps the fingerprints to the physico-chemical properties by deep learning.
We propose a GNN model for predicting the DCN, MON, and RON of (oxygenated) hydrocarbons [9]. The model combines our higher order GNN with a recurrent neural network architecture. Further, it includes a physically meaningful pooling function which adds up contributions of individual atoms to the molecular descriptor. One of the main challenges in this work is the limited availability of experimental data. In order to overcome this difficulty, we study multitask learning, transfer learning, and ensemble learning. The results show competitive performance of the proposed GNN approach compared to state-of-the-art QSPRs making it a promising field for future research. The model and training code is open-source available at
https://git.rwth-aachen.de/avt.svt/public/graph_neural_network_for_fuel_ignition_quality
and a prediction tool is available via a web front-end at http://www.avt.rwth-aachen.de/gnn
[1] Gschwend, D., Soltic, P., Wokaun, A., & Vogel, F. (2019). Review and performance evaluation of fifty alternative liquid fuels for spark-ignition engines. Energy & Fuels, 33(3), 2186-2196.
[2] Dahmen, M., & Marquardt, W. (2015). A novel group contribution method for the prediction of the derived cetane number of oxygenated hydrocarbons. Energy & Fuels, 29(9), 5781-5801.
[3] Kubic Jr, W. L., Jenkins, R. W., Moore, C. M., Semelsberger, T. A., & Sutton, A. D. (2017). Artificial neural network based group contribution method for estimating cetane and octane numbers of hydrocarbons and oxygenated organic compounds. Industrial & Engineering Chemistry Research, 56(42), 12236-12245.
[4] Kessler, T., Sacia, E. R., Bell, A. T., & Mack, J. H. (2017). Artificial neural network based predictions of cetane number for furanic biofuel additives. Fuel, 206, 171-179.
[5] Smolenskii, E. A., Bavykin, V. M., Ryzhov, A. N., Slovokhotova, O. L., Chuvaeva, I. V., & Lapidus, A. L. (2008). Cetane numbers of hydrocarbons: calculations using optimal topological indices. Russian Chemical Bulletin, 57(3), 461-467.
[6] Kipf, T. N., & Welling, M. (2017). Semi-supervised classification with graph convolutional networks. Proceedings of the 5th International Conference on Learning Representations.
[7] Hamilton, W., Ying, R., & Leskovec J. (2017). Inductive Representation Learning on Large Graphs. Proceedings of the 30th Annual Conference on Neural Information Processing Systems, 1024-1034.
[8] Coley, C. W., Jin, W., Rogers, L., Jamison, T. F., Jaakkola, T. S., Green, W. H., Barzilay, R. & Jensen, K. F. (2019). A graph-convolutional neural network model for the prediction of chemical reactivity. Chemical Science, 10(2), 370-377.
[9] Schweidtmann, A. M., Rittig, J. G., König, A., Grohe, M., Mitsos, A., & Dahmen, M. (2020). Graph Neural Networks for Prediction of Fuel Ignition Quality. Submitted for Journal Publication.