2020 Virtual AIChE Annual Meeting
(334e) Enabling Selective Conversion of Biomass Derived Oxygenates to C4-C5 Dienes
Authors
The dependence of our civilization on plastics cannot be overstated â in 2018 alone, the worldwide consumption of plastics exceeded 300 million tons (~40 kg per person on earth). The carbon backbone of plastics is composed of monomeric hydrocarbons like C4 and C5 dienes. Diene market is valued at nearly $4 billion, and alongside steel and ammonia, diene production is integral to the growth of economies. These chemicals (butadiene, pentadienes, and isoprene) are currently produced by the cracking of naphtha fraction of crude oil, where ethylene is the major product. With a recent shift towards using shale-gas (primarily composed of light molecules like methane and ethane) as a cheap alternative to crude oil for ethylene production, refineries are decoupling production routes of ethylene and larger fractions including dienes. Consequently, butadiene production in the US has dropped by ca. 30% since 2007.As demands continue to rise and supplies fall, other on-purpose and possibly renewable routes that are not coupled to ethylene production are required.
One such chemical pathway utilizes the tandem-ring opening dehydration (hereinafter referred to as dehydra-decyclization) of biomass-derived saturated furanic species to produce these dienes (Scheme 1). For example, four-carbon tetrahydrofuran (THF) reacts on the surface of a solid Brønsted acid catalyst to produce butadiene, while five-carbon methylated tetrahydrofuran (2-MTHF) yields linear pentadienes. Limited prior investigations of this chemistry had not rigorously tackled the three fundamental catalyst design challenges, namely assessing: rates (how fast the reaction occurs per active site of the catalyst), diene selectivities (how much of fed-in carbon ends up in desirable product/s), and lifetime (catalyst stability under reaction conditions). In addition, the origin of side products (propene, formaldehyde, and alkylated aromatics), as well as the identity of key reaction intermediates were unknown. The breadth of accessible dienes would be afforded by our ability to prepare numerous substituted THF precursors, and yet it remained unclear how these substituents tune the chemistry of diene formation. Due to this lack of mechanistic knowledge, the maximum diene selectivities attainable in the chemistry had remained low (< 60%); increasing these selectivities would drive down energy-intensive downstream separation costs, and potentially lead to economic feasibility of the overall process. My dissertation has focused on gaining fundamental insights into the dehydra-decyclization of saturated furans to guide a rational search for active, selective, and stable catalysts for this chemistry.
Below, I highlight the results from three independent studies that detail my efforts in i) designing a chemical reactor capable of automated high-throughput catalytic evaluation, followed by ii) detailed kinetic evaluation of THF dehydra-decyclization to expound the catalytic mechanisms and pathways for the chemistry, and finally iii) the design and utilization of a selective and stable class of catalysts exhibiting ~89% diene selectivities at complete furan conversion.
1) Design and implementation of a chemical reactor for automated kinetic evaluation : Detailed mechanistic interrogations on a chemistry are preceded by high-throughput screening of a large number of catalysts to find promising candidates and to establish rate, selectivity, and stability trends to aid this search. Typical laboratory-scale packed bed reactors (PBRs) require constant human monitoring owing to multiple independent process control elements, and separate and non-coupled reaction and separation/quantification components, making them unsuitable for high-throughput studies. Motivated by the ability to perform high fidelity kinetic studies with minimal human oversight, I constructed and implemented a method to integrate a typical PBR within a Gas Chromatograph (GC), enabling the control of reaction parameters directly from the GC circuitry (Scheme 2). After carrying out reactor validation tests (e.g. residence time distributions, isothermal operation, absence of heat and mass-transfer limitations), it was found that typical reaction parameters can be obtained with reasonable accuracy with nearly no manual intervention on this low-cost setup. This automated reactor setup has since helped in the successful screening of promising catalysts for THF dehydra-decyclization (as discussed in 3)).
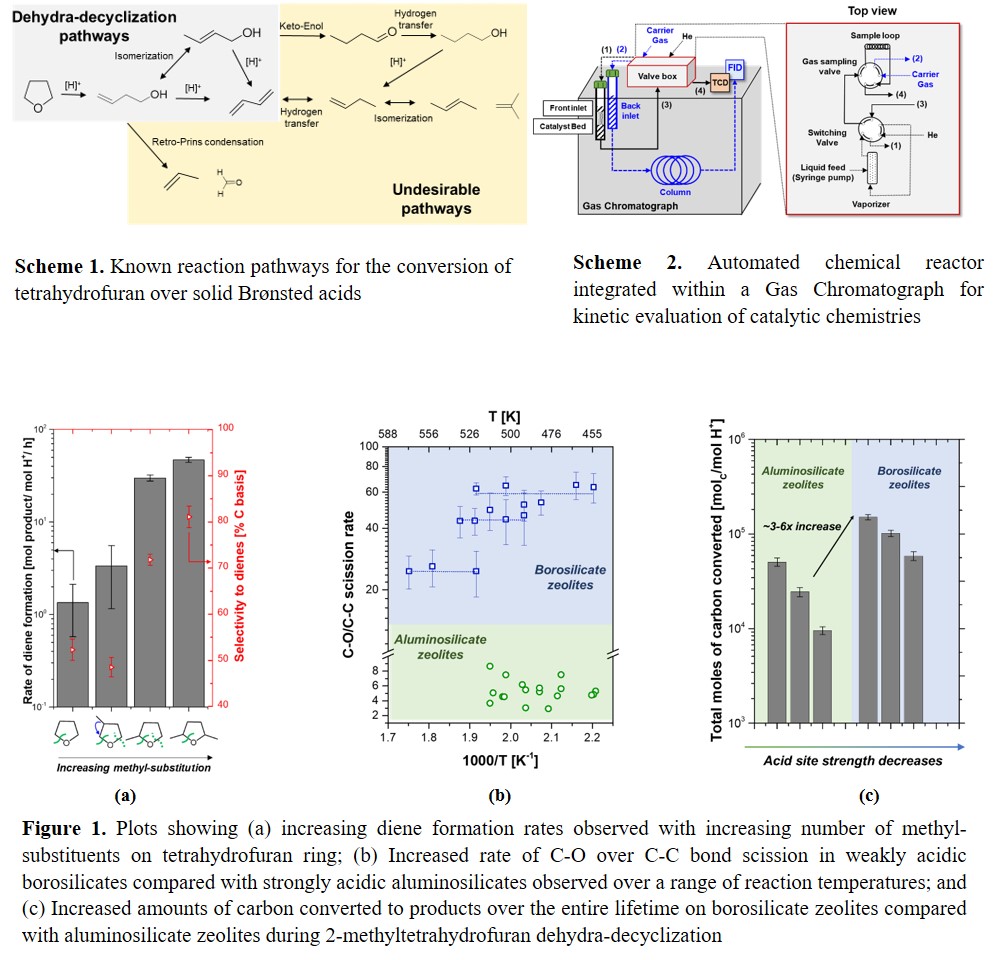
2) Highlighting the mechanisms and pathways for diene formation : We studied THF dehydra-decyclization on an aluminosilicate zeolite H-ZSM-5 by a combination of first-principle calculations, microkinetic modeling, and experimental kinetic measurements. As part of this collaborative effort, my specific contributions to this work were three-fold; i) Experimental kinetic measurements for the chemistry: Assessing THF conversion rates enabled the identification of two key deleterious pathways compromising diene selectivities, namely the fragmentation of THF to propene, and C-C condensation pathways to alkylated aromatics. Furthermore, nearly identical activation energies of diene and propene formation pathways guided the search for a common intermediate formed before their respective rate-limiting steps (RDS); ii) Identification of key reaction intermediates for diene production: Co-processing potential intermediates with THF and observing changes in the evolution of butadiene and propene enabled the unambiguous identification of the key intermediates for both desirable and undesirable pathways in the chemistry. Furthermore, a > 100x higher rate of intermediate conversion vs. THF allowed us to conclude that the dehydra-decyclization RDS preceded the dehydration of these intermediates; iii) Experimental identification of the RDS: C-O bond cleavage leading to the ring-opening of THF was hypothesized to be the kinetic bottleneck as it exhibited a high activation barrier of ~ 49 kcal/mol. By stabilizing the carbenium ion transition state formed during this step by methyl-substitutions on the THF ring, I was able to increase the diene formation rates by > 50x (Figure 1a), experimentally demonstrating the rate-limiting nature of furan ring-opening step for the first time.
3) Enhancing diene selectivities by weakly acidic catalysts : Explicating the mechanisms and pathways of THF dehydra-decyclization cultivated a quantitative understanding about the origin of selectivity; dienes resulted from C-O scission, while the primary side product (propene) originated from C-C fragmentation of THF ring, meaning that the diene selective catalysts should selectively activate C-O over C-C bonds. I hypothesized one possible âknobâ to tune C-O/C-C bond scission rates to be the Brønsted acidic site strength, as the energetics of proton transfer from the weak acid are likely more favorable towards C-O than C-C, owing to its significantly higher proton affinity. Catalytic evaluation of weakly acidic borosilicate zeolites in different surrounding framework environments consistently exhibited ca. 3-30x higher C-O/C-C scission rates than strongly acidic aluminosilicate zeolites (Figure 1b), evincing the inability of weak acids to cleave C-C bonds, and leading to ~10-30% increase in diene selectivities. In addition, borosilicate zeolites converted ~ 3-6x higher amounts of carbon from the feed to dienes during their lifetime than aluminosilicate zeolites (Figure 1c). This remarkable stability was conferred by the inability of these sites to bind and catalyze C-C condensation pathways to alkylated aromatics, which coke up the catalyst and limit their lifetime. The suppression of competing pathways lead to an unprecedented â¼ 89% combined linear C5 dienesâ yield from 2-MTHF, a 30% improvement from the previous best performing catalysts.
In short, my research has provided key mechanistic insights into a sustainable catalytic route to produce commercially high-volume dienes from biomass-derived feed, which in turn, has enabled the discovery of selective and stable catalysts for this chemistry. As our society gradually moves towards renewable fuels and chemicals production, I believe that the results from this dissertation will be helpful in potential integration of this process chemistry with other hydrocarbon production routes within a biorefinery.