(478c) In-Situ Structure Dependent Water Oxidation Activity on Iridium Oxide Single Crystal Surfaces
AIChE Annual Meeting
2022
2022 Annual Meeting
Catalysis and Reaction Engineering Division
Electrocatalysis II: Catalyst and Characterization
Wednesday, November 16, 2022 - 1:06pm to 1:24pm
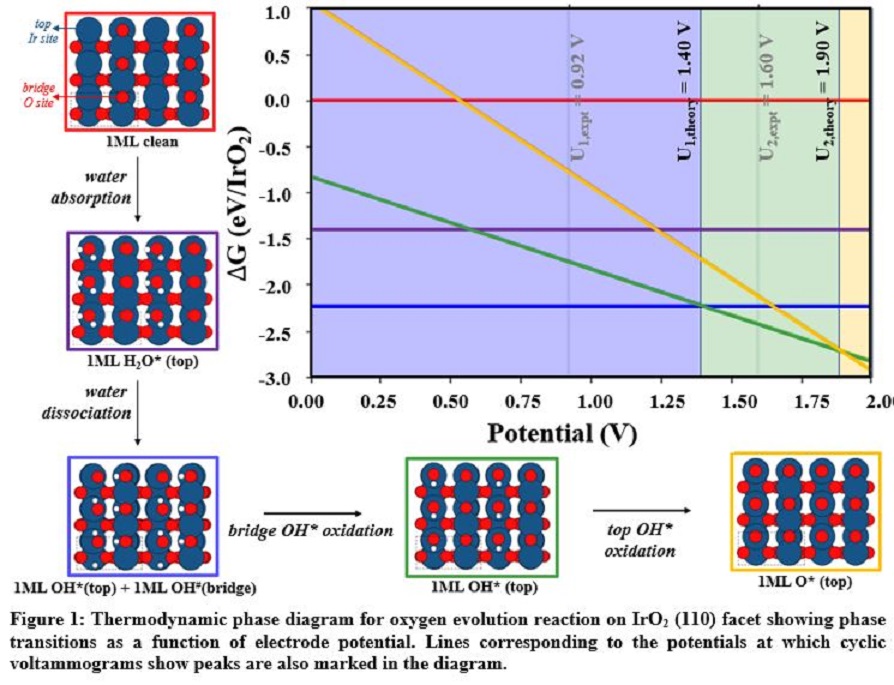
Iridium oxides have been considered as a benchmark catalyst exhibiting high activity for the oxygen evolution reaction (OER) in acidic media. Its structure, however, is debated with phase transformations possible at the surface or in the bulk under oxidizing potentials probing which could be helpful with providing insights into the reasons for its stability loss. In this study, we computationally probe the structural evolution of a single crystal (110) surface of IrO2 from thermodynamic phase diagrams using density functional theory (DFT). We capture co-adsorption effects at high and mixed OER intermediate coverages employing a graph theory algorithm, while assuming solvation corrections for hydrophilic OH* and H2O* intermediates akin to ones reported on metals as a first approximation. Our results indicate that the top iridium sites on the (110) surface are completely covered with water in an aqueous environment. This water undergoes further dissociation forming a monolayer of OH* on the top sites, and protonating lattice oxygen atoms at bridging iridium atoms to form OH# at the bridge positions. Our phase diagram analysis further indicates that that the bridge OH# groups oxidize to give back bridge lattice oxygen atoms at oxidizing potentials of 1.40 V. The top OH* groups, on the other hand, oxidize to top O* groups as the potential is raised to 1.90 V. These computationally predicted phase transition potentials correctly predict the trend of the experimentally observed peaks of 0.92 V and 1.60 V, reported from cyclic voltammograms by Suntivich et al.1 We discuss overprediction from computations in the context of sensitivity to DFT functionals, change in oxidation state evaluated with a Bader charge analysis and DFT+U corrections. We are now explicitly determining coverage dependent solvation corrections using an in-house developed simulated annealing inspired scheme to test its sensitivity to computationally predicted potentials.
- Kuo et al., JACS, 2017